Allen H. Chen, Zachary J. Knepp, Christian A. Guzman, Elizabeth R. Young and Lisa A. Fredin
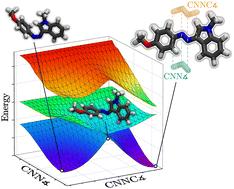
{"title":"Intramolecular subtleties in indole azo dyes revealed by multidimensional potential energy surfaces†","authors":"Allen H. Chen, Zachary J. Knepp, Christian A. Guzman, Elizabeth R. Young and Lisa A. Fredin","doi":"10.1039/D5CP00110B","DOIUrl":null,"url":null,"abstract":"<p >Despite their wide use as molecular photoswitches, the mechanistic photophysics of azo dyes are complex and nuanced, and therefore under-explored. To understand the complex electronic interactions that govern the photoisomerization and thermal reversion of two phenyl-azo-indole dyes that differ by R-sterics near the azo bond, potential energy surfaces that combine the dihedral rotation of the azo bond and the aryl inversion on each side of the azo bond were calculated with density functional theory and time-dependent density functional theory. These multidimensional singlet surfaces provide insights into the correlated rotation and inversion pathways allowing for detailed understanding of both photoisomerization, governed by the excited-state surfaces, and thermal reversion, governed by the ground-state surface, mechanisms to be developed. Large plateaus on the S<small><sub>1</sub></small> surface arise from strong intramolecular interactions between a phenyl substituent and one of the aryl groups, extending the experimental photoisomerization lifetime of the dye with a phenyl R-group by two times over the unsubstituted dye. While one might expect the sterics of the larger phenyl substituent to lead to a slower thermal reversion rate, this was not the case. The thermally accessible <em>meta</em>-stable rotamers of the <em>cis</em>-isomer leads to more reversion pathways and a longer <em>cis</em>-lifetime for the unsubstituted dye, by a factor of four in the experiment. Careful computational mapping of multidimensional potential energy surfaces allows accurate mechanistic understanding for systems with interdependent degrees of freedom between meta-stable states.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 13","pages":" 6430-6437"},"PeriodicalIF":2.9000,"publicationDate":"2025-03-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d5cp00110b?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00110b","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
Despite their wide use as molecular photoswitches, the mechanistic photophysics of azo dyes are complex and nuanced, and therefore under-explored. To understand the complex electronic interactions that govern the photoisomerization and thermal reversion of two phenyl-azo-indole dyes that differ by R-sterics near the azo bond, potential energy surfaces that combine the dihedral rotation of the azo bond and the aryl inversion on each side of the azo bond were calculated with density functional theory and time-dependent density functional theory. These multidimensional singlet surfaces provide insights into the correlated rotation and inversion pathways allowing for detailed understanding of both photoisomerization, governed by the excited-state surfaces, and thermal reversion, governed by the ground-state surface, mechanisms to be developed. Large plateaus on the S1 surface arise from strong intramolecular interactions between a phenyl substituent and one of the aryl groups, extending the experimental photoisomerization lifetime of the dye with a phenyl R-group by two times over the unsubstituted dye. While one might expect the sterics of the larger phenyl substituent to lead to a slower thermal reversion rate, this was not the case. The thermally accessible meta-stable rotamers of the cis-isomer leads to more reversion pathways and a longer cis-lifetime for the unsubstituted dye, by a factor of four in the experiment. Careful computational mapping of multidimensional potential energy surfaces allows accurate mechanistic understanding for systems with interdependent degrees of freedom between meta-stable states.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


