{"title":"[Integrative transcriptomics-metabolomics approach to identify metabolic pathways regulated by glutamine synthetase activity].","authors":"Ting Ling, Jing Shi, Ting-Ze Feng, Shao-Jun Pei, Si-Yi Li, Hai-Long Piao","doi":"10.3724/SP.J.1123.2024.04003","DOIUrl":null,"url":null,"abstract":"<p><p>Glutamine synthetase (GS), the only enzyme responsible for de novo glutamine synthesis, plays a significant role in cancer progression. As an example of the consequences of GS mutations, the R324C variant causes congenital glutamine deficiency, which results in brain abnormalities and neonatal death. However, the influence of GS-deficient mutations on cancer cells remains relatively unexplored. In this study, we investigated the effects of GS and GS-deficient mutations, including R324C and previously unreported K241R, which serve as models for GS inactivation. This study provided intriguing insights into the intricate relationship between GS mutations and cancer cell metabolism. Our findings strongly support recent studies that suggest GS deletion leads to the suppression of diverse signaling cascades associated with glutamine metabolism under glutamine-stripping conditions. The affected processes include DNA synthesis, the citric acid cycle, and reactive oxygen species (ROS) detoxification. This suppression originates from the inherent inability of cells to autonomously synthesize glutamine under glutamine-depleted conditions. As a key source of reduced nitrogen, glutamine is crucial for the formation of purine and pyrimidine bases, which are essential building blocks for DNA synthesis. Furthermore, the citric acid cycle is inhibited by the absence of negatively charged glutamate within the mitochondrial matrix, particularly when glutamine is scarce. This deficiency decreases the flux of <i>α</i>-ketoglutarate (<i>α</i>-KG), a principal driver of the citric acid cycle. Intermediate metabolites of the citric acid cycle directly or indirectly contribute to the generation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, a core component of redox homeostasis. Using the GS_R324C and GS_K241R mutants, we conducted an integrative transcriptomics and metabolomics analysis. The GS mutants with reduced activity activated multiple amino acid biosynthesis pathways, including arginine-proline, glycine-serine-threonine, and alanine-aspartate-glutamate metabolism. This intriguing behavior led us to hypothesize that despite hindrance of the citric acid cycle, abundant intracellular glutamate is redirected through alternative processes, including transamination. Simultaneously, key metabolic enzymes in the amino acid synthesis pathways, such as glutamic-oxaloacetic transaminase 1 (GOT1), glutamic-pyruvic transaminase 2 (GPT2), pyrroline-5-carboxylate reductase 1 (PYCR1), and phosphoserine aminotransferase 1 (PSAT1), exhibited increased mRNA levels. Additionally, GS deficiency appeared to upregulate the expression of glutamine transporters SLC38A2 and SLC1A5. Thus, restricting extracellular amino acids, such as glutamine, induces a stress response while promoting transcription or translation by a select group of genes, thereby facilitating cellular adaptation. However, similar to GS_WT, both GS_R324C and GS_K241R were modulated by glutamine treatment. Among GS-activity-dependent behaviors, the increased expression of numerous aminoacyl-tRNA synthetases (ARSs), which are critical for aminoacyl-tRNA biosynthesis, remains poorly understood. Most ARS-encoding genes are transcriptionally induced by activating transcription factor 4 (ATF4), the expression of which increases under oxidative stress, endoplasmic reticulum stress, hypoxia, and amino acid limitation. In GS-deficient cells, the increased expression of ATF4 was accompanied by pronounced stress caused by glutamine starvation. Thus, ARS upregulation may predominantly arise from increased ATF4 expression in GS-deficient cells. Additionally, transcriptomic analysis revealed the differential expression of specific genes, regardless of GS activity, suggesting that GS is involved in various processes other than glutamine synthesis, including angiogenesis. Although our omics study was limited to H1299 cells, in subsequent experiments, we validated our findings using additional cell lines, including Hepa1-6 and LN-229. To attain a more comprehensive understanding of the impact of the newly identified GS_K241R mutant, our investigation should be extended to various cell types and mouse models. In summary, we identified and investigated GS-deficient mutations in cancer cells and conducted an integrative transcriptomics-metabolomics analysis with comparisons to wild-type GS. This comprehensive approach provided crucial insights into the intricate pathways modulated by GS activity. Our findings advance the understanding of how GS functions in the context of reprogrammed cellular metabolism, particularly during glutamine deprivation. The altered metabolism triggered by elevated glutamate levels arising from GS mutations highlights the remarkable plasticity of cancer cell metabolism. Notably, considering the increasing research focus on GS as a potential therapeutic target in various cancer types, the findings of this study could provide innovative perspectives for drug development and the formulation of clinical treatment strategies.</p>","PeriodicalId":101336,"journal":{"name":"Se pu = Chinese journal of chromatography","volume":"43 3","pages":"207-219"},"PeriodicalIF":0.0000,"publicationDate":"2025-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11883535/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Se pu = Chinese journal of chromatography","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3724/SP.J.1123.2024.04003","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
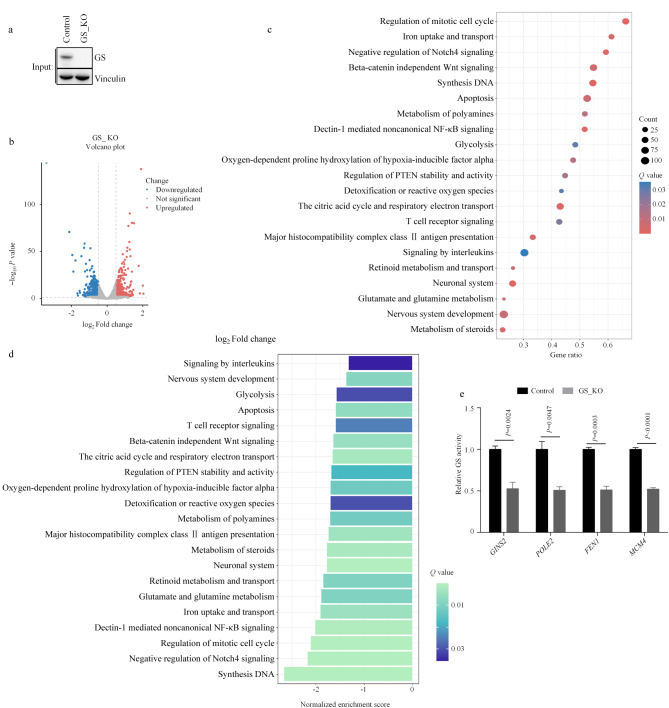
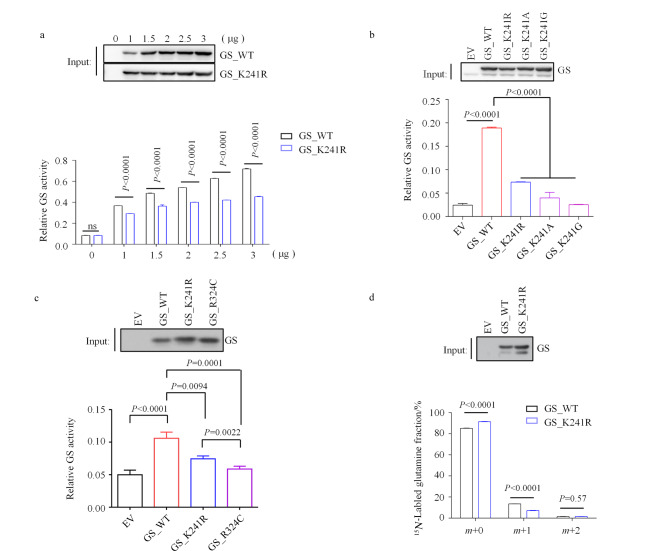
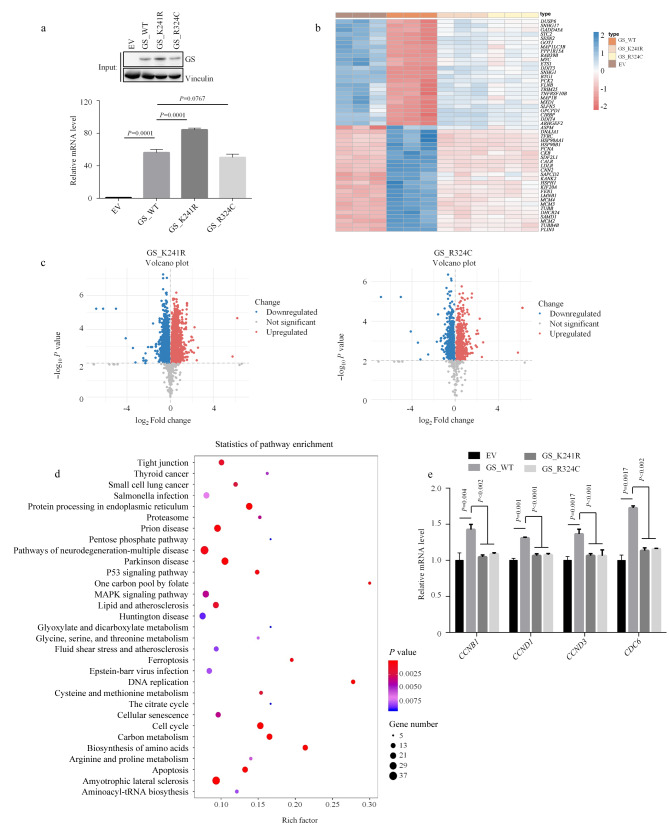
Abstract
Glutamine synthetase (GS), the only enzyme responsible for de novo glutamine synthesis, plays a significant role in cancer progression. As an example of the consequences of GS mutations, the R324C variant causes congenital glutamine deficiency, which results in brain abnormalities and neonatal death. However, the influence of GS-deficient mutations on cancer cells remains relatively unexplored. In this study, we investigated the effects of GS and GS-deficient mutations, including R324C and previously unreported K241R, which serve as models for GS inactivation. This study provided intriguing insights into the intricate relationship between GS mutations and cancer cell metabolism. Our findings strongly support recent studies that suggest GS deletion leads to the suppression of diverse signaling cascades associated with glutamine metabolism under glutamine-stripping conditions. The affected processes include DNA synthesis, the citric acid cycle, and reactive oxygen species (ROS) detoxification. This suppression originates from the inherent inability of cells to autonomously synthesize glutamine under glutamine-depleted conditions. As a key source of reduced nitrogen, glutamine is crucial for the formation of purine and pyrimidine bases, which are essential building blocks for DNA synthesis. Furthermore, the citric acid cycle is inhibited by the absence of negatively charged glutamate within the mitochondrial matrix, particularly when glutamine is scarce. This deficiency decreases the flux of α-ketoglutarate (α-KG), a principal driver of the citric acid cycle. Intermediate metabolites of the citric acid cycle directly or indirectly contribute to the generation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, a core component of redox homeostasis. Using the GS_R324C and GS_K241R mutants, we conducted an integrative transcriptomics and metabolomics analysis. The GS mutants with reduced activity activated multiple amino acid biosynthesis pathways, including arginine-proline, glycine-serine-threonine, and alanine-aspartate-glutamate metabolism. This intriguing behavior led us to hypothesize that despite hindrance of the citric acid cycle, abundant intracellular glutamate is redirected through alternative processes, including transamination. Simultaneously, key metabolic enzymes in the amino acid synthesis pathways, such as glutamic-oxaloacetic transaminase 1 (GOT1), glutamic-pyruvic transaminase 2 (GPT2), pyrroline-5-carboxylate reductase 1 (PYCR1), and phosphoserine aminotransferase 1 (PSAT1), exhibited increased mRNA levels. Additionally, GS deficiency appeared to upregulate the expression of glutamine transporters SLC38A2 and SLC1A5. Thus, restricting extracellular amino acids, such as glutamine, induces a stress response while promoting transcription or translation by a select group of genes, thereby facilitating cellular adaptation. However, similar to GS_WT, both GS_R324C and GS_K241R were modulated by glutamine treatment. Among GS-activity-dependent behaviors, the increased expression of numerous aminoacyl-tRNA synthetases (ARSs), which are critical for aminoacyl-tRNA biosynthesis, remains poorly understood. Most ARS-encoding genes are transcriptionally induced by activating transcription factor 4 (ATF4), the expression of which increases under oxidative stress, endoplasmic reticulum stress, hypoxia, and amino acid limitation. In GS-deficient cells, the increased expression of ATF4 was accompanied by pronounced stress caused by glutamine starvation. Thus, ARS upregulation may predominantly arise from increased ATF4 expression in GS-deficient cells. Additionally, transcriptomic analysis revealed the differential expression of specific genes, regardless of GS activity, suggesting that GS is involved in various processes other than glutamine synthesis, including angiogenesis. Although our omics study was limited to H1299 cells, in subsequent experiments, we validated our findings using additional cell lines, including Hepa1-6 and LN-229. To attain a more comprehensive understanding of the impact of the newly identified GS_K241R mutant, our investigation should be extended to various cell types and mouse models. In summary, we identified and investigated GS-deficient mutations in cancer cells and conducted an integrative transcriptomics-metabolomics analysis with comparisons to wild-type GS. This comprehensive approach provided crucial insights into the intricate pathways modulated by GS activity. Our findings advance the understanding of how GS functions in the context of reprogrammed cellular metabolism, particularly during glutamine deprivation. The altered metabolism triggered by elevated glutamate levels arising from GS mutations highlights the remarkable plasticity of cancer cell metabolism. Notably, considering the increasing research focus on GS as a potential therapeutic target in various cancer types, the findings of this study could provide innovative perspectives for drug development and the formulation of clinical treatment strategies.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


