Limits of BOSS DFT: O2 + Al(111) Dynamics on a Screened Hybrid Van der Waals DFT Potential Energy Surface
IF 3.3
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
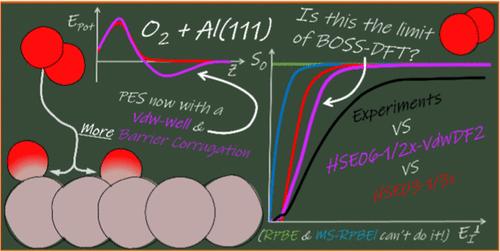
The activated dissociative chemisorption (DC) of O2 on Al(111) is a thoroughly studied benchmark system for oxygen–metal interactions. However, research based on density functional theory (DFT) has not yet been able to accurately determine the electronic structure, and theory as a whole has so far been unable to reproduce measured sticking probabilities with chemical accuracy. Previous work has argued that this is likely due to the inability of DFT at the generalized gradient approximation (GGA) level to describe the barriers to DC of O2 on Al(111) correctly. The argument is that the most commonly applied electronic structure approach in surface science, which involves the use of GGA-DFT, yields too low reaction barriers for the DC of O2 on Al(111). Moreover, it seems that GGAs will generally fail to accurately predict barriers for systems with low charge transfer energy, i.e., systems for which charge transfer from metal to molecule at the transition state is likely. Subsequent work on both O2 + Al(111) and O2 + Cu(111) has suggested that screened hybrid density functionals (DF) yield more accurate barrier heights for DC on metal surfaces. However, so far the use of only a screened hybrid DF was not enough to ensure a highly accurate description for O2 + Al(111). Even though the onset of the sticking probability (S0) curve was correctly described, the slope, or width, of the curve was not. The use of a nonlocal correlation DF combined with an increased fraction of exact exchange in the screened hybrid exchange DF was believed to further improve the description of the electronic structure by increasing the energetic corrugation of the barrier. This approach was assumed to increase the width of the sticking curve without lowering the incidence energy for the reaction onset, thus reducing the slope of the sticking curve. To test this, we present quasi-classical trajectory (QCT) calculations on the O2 + Al(111) system based on a potential energy surface (PES) computed with the HSE06-1/2x-VdWDF2 screened hybrid van der Waals DF, using the Born–Oppenheimer static surface (BOSS) model. The resulting PES shows the presence of shallow van der Waals wells in the entrance channel. Furthermore, the barriers to DC show a slightly higher energetic corrugation than the previously used HSE03-1/3x screened hybrid DF, although most differences are smaller than 1 kcal/mol. These minor alterations in the PES with respect to previous work mean that the S0 computed for O2 + Al(111) using the HSE06-1/2x-VdWDF2 DF are somewhat improved over the previous results. Specifically, the onset of the S0 curve is now somewhat better described and the curve is broadened a little compared to the HSE03-1/3x description. These results, in combination with previous studies, imply that future electronic structure methods would need to provide larger changes in the PES, or a different dynamical model would need to be used to bring theory in better agreement with the experiment. Moreover, future higher-level theory also needs to address the currently very demanding computational costs of screened hybrid plane-wave-DFT for molecule-metal interactions.
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


