{"title":"MolEpidPred: a novel computational tool for the molecular epidemiology of foot-and-mouth disease virus using VP1 nucleotide sequence data.","authors":"Samarendra Das, Utkal Nayak, Soumen Pal, Saravanan Subramaniam","doi":"10.1093/bfgp/elaf001","DOIUrl":null,"url":null,"abstract":"<p><p>Molecular epidemiology of Foot-and-mouth disease (FMD) is crucial to implement its control strategies including vaccination and containment, which primarily deals with knowing serotype, topotype, and lineage of the virus. The existing approaches including serotyping are biological in nature, which are time-consuming and risky due to live virus handling. Thus, novel computational tools are highly required for large-scale molecular epidemiology of the FMD virus. This study reported a comprehensive computational tool for FMD molecular epidemiology. Ten learning algorithms were initially evaluated on cross-validated and ten independent secondary datasets for serotype prediction using sequence-based features through accuracy, sensitivity and 14 other metrics. Next, best performing algorithms, with higher serotype predictive accuracies, were evaluated for topotype and lineage prediction using cross-validation. These algorithms are implemented in the computational tool. Then, performance of the developed approach was assessed on five independent secondary datasets, never seen before, and primary experimental data. Our cross-validated and independent evaluation of learning algorithms for serotype prediction revealed that support vector machine, random forest, XGBoost, and AdaBoost algorithms outperformed others. Then, these four algorithms were evaluated for topotype and lineage prediction, which achieved accuracy ≥96% and precision ≥95% on cross-validated data. These algorithms are implemented in the web-server (https://nifmd-bbf.icar.gov.in/MolEpidPred), which allows rapid molecular epidemiology of FMD virus. The independent validation of the MolEpidPred observed accuracies ≥98%, ≥90%, and ≥ 80% for serotype, topotype, and lineage prediction, respectively. On wet-lab data, the MolEpidPred tool provided results in fewer seconds and achieved accuracies of 100%, 100%, and 96% for serotype, topotype, and lineage prediction, respectively, when benchmarked with phylogenetic analysis. MolEpidPred tool provides an innovative platform for large-scale molecular epidemiology of FMD virus, which is crucial for tracking FMD virus infection and implementing control program.</p>","PeriodicalId":55323,"journal":{"name":"Briefings in Functional Genomics","volume":"24 ","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-01-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11881699/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Briefings in Functional Genomics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bfgp/elaf001","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
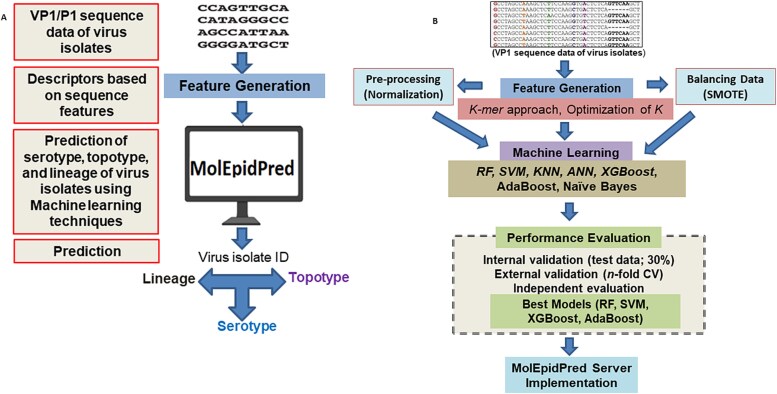
Molecular epidemiology of Foot-and-mouth disease (FMD) is crucial to implement its control strategies including vaccination and containment, which primarily deals with knowing serotype, topotype, and lineage of the virus. The existing approaches including serotyping are biological in nature, which are time-consuming and risky due to live virus handling. Thus, novel computational tools are highly required for large-scale molecular epidemiology of the FMD virus. This study reported a comprehensive computational tool for FMD molecular epidemiology. Ten learning algorithms were initially evaluated on cross-validated and ten independent secondary datasets for serotype prediction using sequence-based features through accuracy, sensitivity and 14 other metrics. Next, best performing algorithms, with higher serotype predictive accuracies, were evaluated for topotype and lineage prediction using cross-validation. These algorithms are implemented in the computational tool. Then, performance of the developed approach was assessed on five independent secondary datasets, never seen before, and primary experimental data. Our cross-validated and independent evaluation of learning algorithms for serotype prediction revealed that support vector machine, random forest, XGBoost, and AdaBoost algorithms outperformed others. Then, these four algorithms were evaluated for topotype and lineage prediction, which achieved accuracy ≥96% and precision ≥95% on cross-validated data. These algorithms are implemented in the web-server (https://nifmd-bbf.icar.gov.in/MolEpidPred), which allows rapid molecular epidemiology of FMD virus. The independent validation of the MolEpidPred observed accuracies ≥98%, ≥90%, and ≥ 80% for serotype, topotype, and lineage prediction, respectively. On wet-lab data, the MolEpidPred tool provided results in fewer seconds and achieved accuracies of 100%, 100%, and 96% for serotype, topotype, and lineage prediction, respectively, when benchmarked with phylogenetic analysis. MolEpidPred tool provides an innovative platform for large-scale molecular epidemiology of FMD virus, which is crucial for tracking FMD virus infection and implementing control program.
期刊介绍:
Briefings in Functional Genomics publishes high quality peer reviewed articles that focus on the use, development or exploitation of genomic approaches, and their application to all areas of biological research. As well as exploring thematic areas where these techniques and protocols are being used, articles review the impact that these approaches have had, or are likely to have, on their field. Subjects covered by the Journal include but are not restricted to: the identification and functional characterisation of coding and non-coding features in genomes, microarray technologies, gene expression profiling, next generation sequencing, pharmacogenomics, phenomics, SNP technologies, transgenic systems, mutation screens and genotyping. Articles range in scope and depth from the introductory level to specific details of protocols and analyses, encompassing bacterial, fungal, plant, animal and human data.
The editorial board welcome the submission of review articles for publication. Essential criteria for the publication of papers is that they do not contain primary data, and that they are high quality, clearly written review articles which provide a balanced, highly informative and up to date perspective to researchers in the field of functional genomics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


