Chris Jennings-Shaffer, David H Rich, Matthew Macaulay, Michael D Karcher, Tanvi Ganapathy, Shosuke Kiami, Anna Kooperberg, Cheng Zhang, Marc A Suchard, Frederick A Matsen
{"title":"Finding high posterior density phylogenies by systematically extending a directed acyclic graph.","authors":"Chris Jennings-Shaffer, David H Rich, Matthew Macaulay, Michael D Karcher, Tanvi Ganapathy, Shosuke Kiami, Anna Kooperberg, Cheng Zhang, Marc A Suchard, Frederick A Matsen","doi":"10.1186/s13015-025-00273-x","DOIUrl":null,"url":null,"abstract":"<p><p>Bayesian phylogenetics typically estimates a posterior distribution, or aspects thereof, using Markov chain Monte Carlo methods. These methods integrate over tree space by applying local rearrangements to move a tree through its space as a random walk. Previous work explored the possibility of replacing this random walk with a systematic search, but was quickly overwhelmed by the large number of probable trees in the posterior distribution. In this paper we develop methods to sidestep this problem using a recently introduced structure called the subsplit directed acyclic graph (sDAG). This structure can represent many trees at once, and local rearrangements of trees translate to methods of enlarging the sDAG. Here we propose two methods of introducing, ranking, and selecting local rearrangements on sDAGs to produce a collection of trees with high posterior density. One of these methods successfully recovers the set of high posterior density trees across a range of data sets. However, we find that a simpler strategy of aggregating trees into an sDAG in fact is computationally faster and returns a higher fraction of probable trees.</p>","PeriodicalId":50823,"journal":{"name":"Algorithms for Molecular Biology","volume":"20 1","pages":"2"},"PeriodicalIF":1.7000,"publicationDate":"2025-02-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11869616/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Algorithms for Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13015-025-00273-x","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
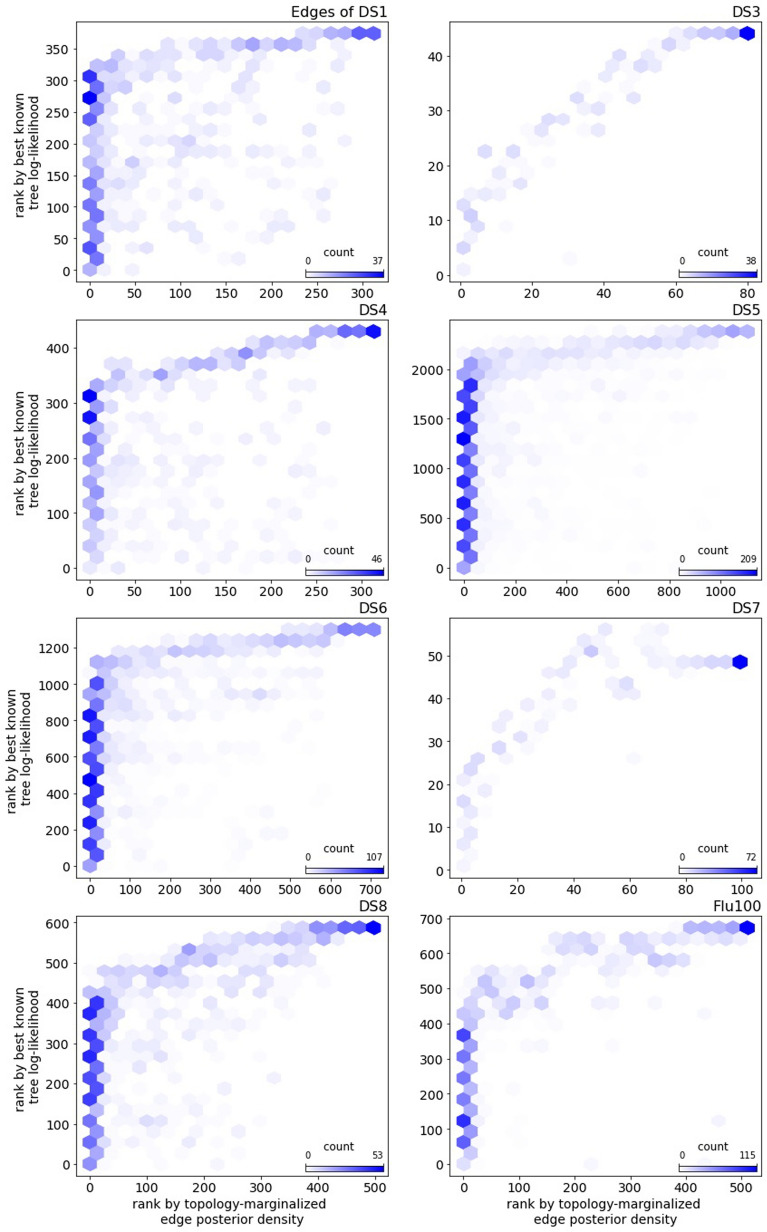
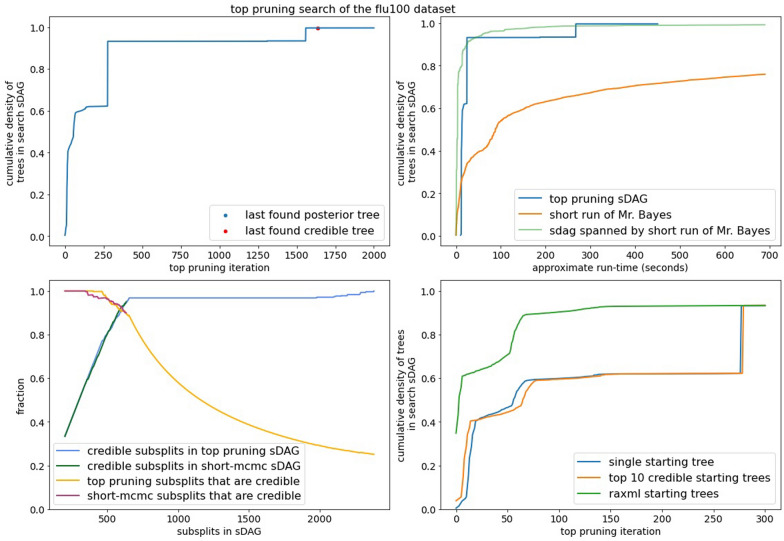
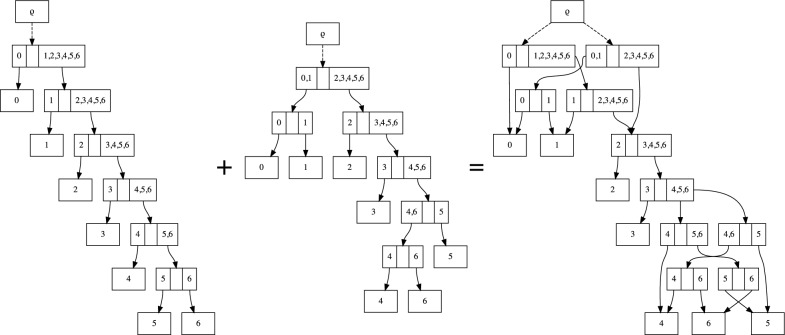
Bayesian phylogenetics typically estimates a posterior distribution, or aspects thereof, using Markov chain Monte Carlo methods. These methods integrate over tree space by applying local rearrangements to move a tree through its space as a random walk. Previous work explored the possibility of replacing this random walk with a systematic search, but was quickly overwhelmed by the large number of probable trees in the posterior distribution. In this paper we develop methods to sidestep this problem using a recently introduced structure called the subsplit directed acyclic graph (sDAG). This structure can represent many trees at once, and local rearrangements of trees translate to methods of enlarging the sDAG. Here we propose two methods of introducing, ranking, and selecting local rearrangements on sDAGs to produce a collection of trees with high posterior density. One of these methods successfully recovers the set of high posterior density trees across a range of data sets. However, we find that a simpler strategy of aggregating trees into an sDAG in fact is computationally faster and returns a higher fraction of probable trees.
期刊介绍:
Algorithms for Molecular Biology publishes articles on novel algorithms for biological sequence and structure analysis, phylogeny reconstruction, and combinatorial algorithms and machine learning.
Areas of interest include but are not limited to: algorithms for RNA and protein structure analysis, gene prediction and genome analysis, comparative sequence analysis and alignment, phylogeny, gene expression, machine learning, and combinatorial algorithms.
Where appropriate, manuscripts should describe applications to real-world data. However, pure algorithm papers are also welcome if future applications to biological data are to be expected, or if they address complexity or approximation issues of novel computational problems in molecular biology. Articles about novel software tools will be considered for publication if they contain some algorithmically interesting aspects.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


