Unraveling the Mechanisms of Li+-Ion Adsorption and Migration on Graphyne and Its BN Analogs
IF 3.3
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
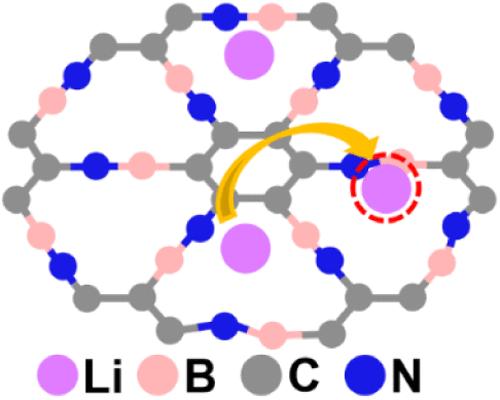
Li-ion batteries are prevalent energy storage systems and are widely utilized in portable devices and electric vehicles. The advent of all-solid-state lithium-ion batteries has recently garnered significant interest due to their enhanced safety profiles. Two-dimensional (2D) materials, such as graphyne (GY) and graphdiyne (GDY), are anticipated to possess a high ion capacity, making them strong candidates for anode materials. Herein, density functional theory (DFT) calculations are performed to explore the adsorption and migration of Li+ ions on the surfaces of GY and its BN analogs (BNyne) that feature –C≡C– linked triangular holes and hexagonal rings (Scheme 1). Our findings reveal that the orbital energy and atomic radius differences among C, B, and N atoms, the polarization of B–N bonds, and the associated charge transfer are the main factors dictating the geometries, electronic structures, and stabilities. Li+-ion adsorption preferentially occurs at a distance of 0.7–1.0 Å above the centers of the triangular holes, with the strongest adsorption occurring on the BNyne-C and the weakest adsorption on BNyne-BN, due to different electrostatic interactions. The migration mechanism involves Li+ ion migrating from the center of one triangular hole to another with an activation barrier of ∼15 kcal/mol. Additional Li+ adsorption at the triangular hole can promote migration due to the reduced adsorption energy. The migration through the benzene-like hexagonal ring is less favored. Notably, among the systems studied, BNyne-BN demonstrates the lowest energy barriers for both migration pathways, indicating superior mobility and rendering it a promising material for potential applications in all-solid-state lithium-ion batteries.
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


