{"title":"The FHL1 myopathy spectrum revisited: a literature review and report of two new patients.","authors":"Maria Caputo, Benedikt Schoser","doi":"10.36185/2532-1900-604","DOIUrl":null,"url":null,"abstract":"<p><strong>Objectives: </strong>Mutations in the FHL1 gene have been associated with a diverse spectrum of X-linked diseases affecting skeletal and cardiac muscle. Six clinically distinct human myopathies can be recognized, including reducing body myopathy (RBM), X-linked dominant scapuloperoneal myopathy (SPM), X-linked myopathy with postural muscle atrophy (XMPMA), rigid spine syndrome (RSS), hypertrophic cardiomyopathy (HCM) and type 6 Emery- Dreifuss muscular dystrophy (EDMD). The core features of all described FHL1opathies are mostly scapuloperoneal muscle weakness, rigid spine, cardiac involvement, and cytoplasmic bodies in the muscle biopsy.</p><p><strong>Methods: </strong>We systematically reviewed the medical literature between the years 2000 and 2024 regarding the phenotype and genotype description of FHL1-associated myopathies.</p><p><strong>Results: </strong>Here, we report two novel patients presenting with an X-linked myopathy with postural muscle atrophy (XMPMA) caused by the c.672 C > G FHL1 gene mutation.</p><p><strong>Conclusion: </strong>When encountering these features in a patient, one may consider screening for an FHL1 mutation. The course ranges from a severe fatal course with early onset to very mild features with late onset. Once a dystrophinopathy has been excluded, increased CK values in male subjects with possible X-linked inheritance should always trigger FHL1 gene screening.</p>","PeriodicalId":93851,"journal":{"name":"Acta myologica : myopathies and cardiomyopathies : official journal of the Mediterranean Society of Myology","volume":"43 4","pages":"123-129"},"PeriodicalIF":0.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11978423/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta myologica : myopathies and cardiomyopathies : official journal of the Mediterranean Society of Myology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.36185/2532-1900-604","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Objectives: Mutations in the FHL1 gene have been associated with a diverse spectrum of X-linked diseases affecting skeletal and cardiac muscle. Six clinically distinct human myopathies can be recognized, including reducing body myopathy (RBM), X-linked dominant scapuloperoneal myopathy (SPM), X-linked myopathy with postural muscle atrophy (XMPMA), rigid spine syndrome (RSS), hypertrophic cardiomyopathy (HCM) and type 6 Emery- Dreifuss muscular dystrophy (EDMD). The core features of all described FHL1opathies are mostly scapuloperoneal muscle weakness, rigid spine, cardiac involvement, and cytoplasmic bodies in the muscle biopsy.
Methods: We systematically reviewed the medical literature between the years 2000 and 2024 regarding the phenotype and genotype description of FHL1-associated myopathies.
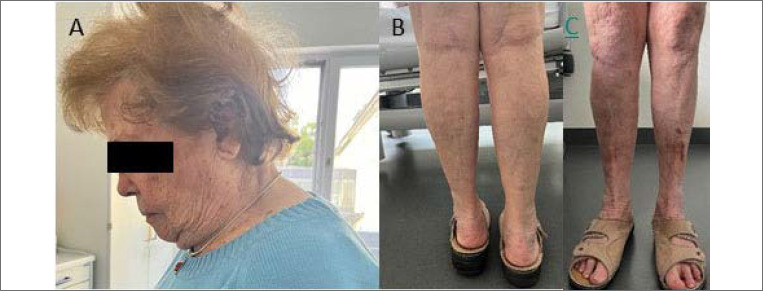
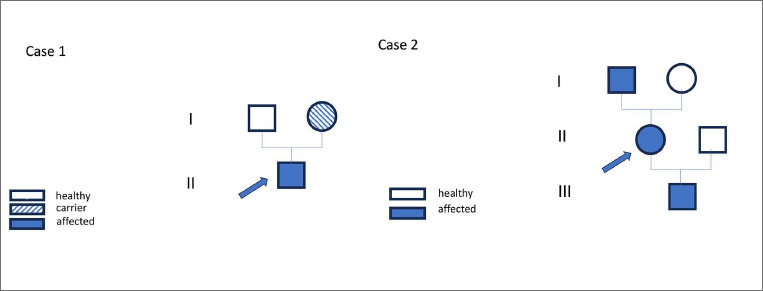
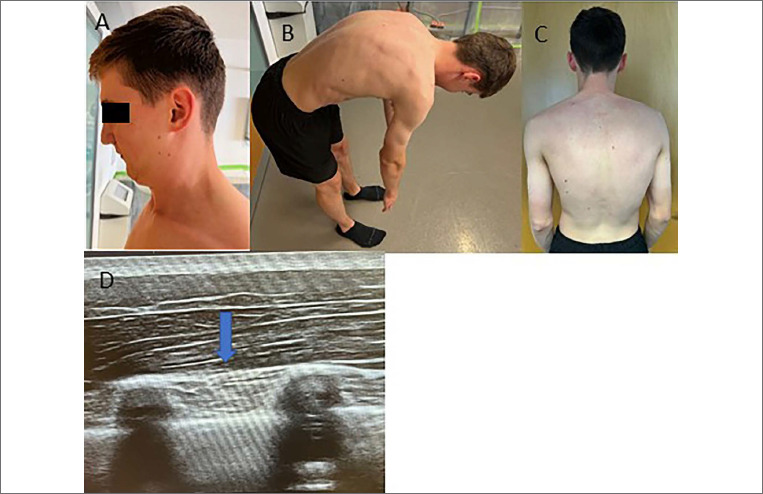
Results: Here, we report two novel patients presenting with an X-linked myopathy with postural muscle atrophy (XMPMA) caused by the c.672 C > G FHL1 gene mutation.
Conclusion: When encountering these features in a patient, one may consider screening for an FHL1 mutation. The course ranges from a severe fatal course with early onset to very mild features with late onset. Once a dystrophinopathy has been excluded, increased CK values in male subjects with possible X-linked inheritance should always trigger FHL1 gene screening.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


