Vinod Rajasingam, Donald P Peter Craig, Yun Hwang, Laveniya Satgunaseelan, Michael Buckland, Rodrigo Tomazini Martins
{"title":"Creutzfeldt-Jakob disease mimicking limbic encephalitis as a cause of rapid neurological deterioration.","authors":"Vinod Rajasingam, Donald P Peter Craig, Yun Hwang, Laveniya Satgunaseelan, Michael Buckland, Rodrigo Tomazini Martins","doi":"10.1136/bmjno-2024-000891","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>A broad range of inflammatory and neurodegenerative conditions manifest with progressive cognitive and behavioural changes. A diagnostic challenge is the differentiation of limbic encephalitis (LE) from Creutzfeldt-Jakob disease (CJD). LE and CJD are distinct neurological conditions with distinct variations in their clinical course, with overlapping clinical presentations. LE can be subdivided into autoimmune paraneoplastic and non-paraneoplastic subtypes, under the umbrella of autoimmune LE. CJD is the most prevalent form of human prion disease and the subtype sporadic CJD (sCJD) the most common.</p><p><strong>Case presentation: </strong>This case study presents a 68-year-old man with a 6-week history of progressive cognitive decline and behavioural changes, ultimately leading to a dire clinical state. The initial symptoms included confusion, intermittent headaches and episodes of aggression towards his wife, preceded by 2 weeks of visual hallucinations. On examination, the patient displayed an ataxic gait, with signs of cerebellar dysfunction. The clinical course evolved, marked by myoclonic jerks, culminating in a decline in both his Glasgow Coma Scale (GCS) score and overall clinical status.</p><p><strong>Conclusion: </strong>The patient's rapidly deteriorating condition over 6 weeks was thought to be too rapid for sCJD, and the patient was treated initially as an LS. However, post-mortem biopsy findings confirmed CJD. Asymmetric periodic discharges on EEG, asymmetric neuroimaging changes and the manifestation of psychiatric symptoms should not preclude the diagnosis of sCJD. This case highlights the importance of recognising the potential rapid deterioration of sCJD, which would alert clinicians to earlier diagnosis and management.</p>","PeriodicalId":52754,"journal":{"name":"BMJ Neurology Open","volume":"7 1","pages":"e000891"},"PeriodicalIF":2.4000,"publicationDate":"2025-02-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11865761/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMJ Neurology Open","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1136/bmjno-2024-000891","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: A broad range of inflammatory and neurodegenerative conditions manifest with progressive cognitive and behavioural changes. A diagnostic challenge is the differentiation of limbic encephalitis (LE) from Creutzfeldt-Jakob disease (CJD). LE and CJD are distinct neurological conditions with distinct variations in their clinical course, with overlapping clinical presentations. LE can be subdivided into autoimmune paraneoplastic and non-paraneoplastic subtypes, under the umbrella of autoimmune LE. CJD is the most prevalent form of human prion disease and the subtype sporadic CJD (sCJD) the most common.
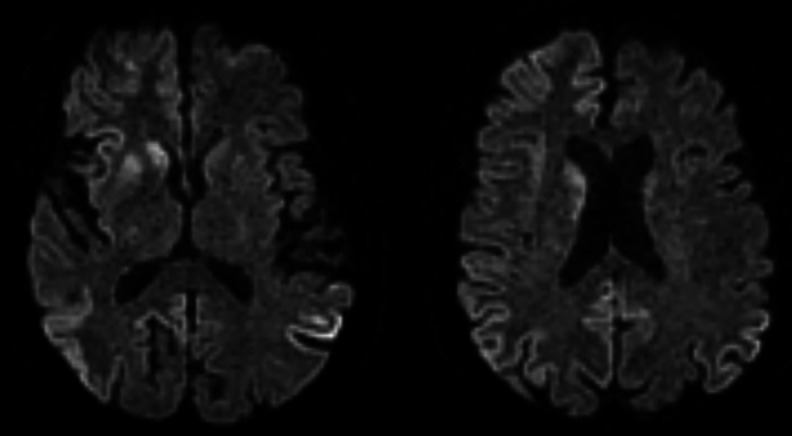
Case presentation: This case study presents a 68-year-old man with a 6-week history of progressive cognitive decline and behavioural changes, ultimately leading to a dire clinical state. The initial symptoms included confusion, intermittent headaches and episodes of aggression towards his wife, preceded by 2 weeks of visual hallucinations. On examination, the patient displayed an ataxic gait, with signs of cerebellar dysfunction. The clinical course evolved, marked by myoclonic jerks, culminating in a decline in both his Glasgow Coma Scale (GCS) score and overall clinical status.
Conclusion: The patient's rapidly deteriorating condition over 6 weeks was thought to be too rapid for sCJD, and the patient was treated initially as an LS. However, post-mortem biopsy findings confirmed CJD. Asymmetric periodic discharges on EEG, asymmetric neuroimaging changes and the manifestation of psychiatric symptoms should not preclude the diagnosis of sCJD. This case highlights the importance of recognising the potential rapid deterioration of sCJD, which would alert clinicians to earlier diagnosis and management.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


