Proton affinities of aldehyde molecules determined from the forward and backward gas-phase proton transfer reactions in a selected ion flow-drift tube†
Maroua Omezzine Gnioua, Anatolii Spesyvyi and Patrik Španěl
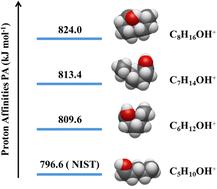
{"title":"Proton affinities of aldehyde molecules determined from the forward and backward gas-phase proton transfer reactions in a selected ion flow-drift tube†","authors":"Maroua Omezzine Gnioua, Anatolii Spesyvyi and Patrik Španěl","doi":"10.1039/D5CP00328H","DOIUrl":null,"url":null,"abstract":"<p >Proton affinity (PA) and gas-phase basicity (GB) are important thermodynamic properties that provide insights into ion–molecule interactions. Aldehydes play a significant role in biology, the environment, and industry, but their PAs remain unknown for those with more than 5 C atoms. This study aims to experimentally determine PAs and GBs of hexanal, heptanal, and octanal using pentanal as a reference. A selected ion flow drift tube (SIFDT) was used to study proton transfer reactions among all possible combinations of protonated and neutral molecules from this set. Rate coefficients (<em>k</em>), equilibrium constants (<em>K</em>), and effective temperatures (<em>T</em><small><sub>eff</sub></small>) were used to calculate Gibbs free energy changes (Δ<em>G</em>) and enthalpy changes (Δ<em>H</em>). PAs and GBs were then determined relative to the known values of pentanal. Experimental PAs were found to increase with aldehyde chain length: pentanal 796.6 kJ mol<small><sup>−1</sup></small> < hexanal 809.6 kJ mol<small><sup>−1</sup></small> < heptanal 813.4 kJ mol<small><sup>−1</sup></small> < octanal 824.0 kJ mol<small><sup>−1</sup></small>. Theoretical enthalpies and entropies were obtained <em>via</em> density functional theory (DFT) B3LYP/6-311++G(d,p) with D4 dispersion correction for both open and bent protonated structures, allowing comparison with experimental data. The theoretical calculations for open structures underestimate the observed PAs, while the bent structures align more closely with experimental trends, indicating that larger protonated aldehydes may have bent and cyclic shapes. These findings contribute to bridging the gaps in knowledge about protonated aldehydes, providing a better understanding of their ion chemistry.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 13","pages":" 6646-6655"},"PeriodicalIF":2.9000,"publicationDate":"2025-02-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d5cp00328h?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00328h","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
Proton affinity (PA) and gas-phase basicity (GB) are important thermodynamic properties that provide insights into ion–molecule interactions. Aldehydes play a significant role in biology, the environment, and industry, but their PAs remain unknown for those with more than 5 C atoms. This study aims to experimentally determine PAs and GBs of hexanal, heptanal, and octanal using pentanal as a reference. A selected ion flow drift tube (SIFDT) was used to study proton transfer reactions among all possible combinations of protonated and neutral molecules from this set. Rate coefficients (k), equilibrium constants (K), and effective temperatures (Teff) were used to calculate Gibbs free energy changes (ΔG) and enthalpy changes (ΔH). PAs and GBs were then determined relative to the known values of pentanal. Experimental PAs were found to increase with aldehyde chain length: pentanal 796.6 kJ mol−1 < hexanal 809.6 kJ mol−1 < heptanal 813.4 kJ mol−1 < octanal 824.0 kJ mol−1. Theoretical enthalpies and entropies were obtained via density functional theory (DFT) B3LYP/6-311++G(d,p) with D4 dispersion correction for both open and bent protonated structures, allowing comparison with experimental data. The theoretical calculations for open structures underestimate the observed PAs, while the bent structures align more closely with experimental trends, indicating that larger protonated aldehydes may have bent and cyclic shapes. These findings contribute to bridging the gaps in knowledge about protonated aldehydes, providing a better understanding of their ion chemistry.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


