Tian-Qi Zhang, Ji-Dong Cai, Cong Li, Yun Xu, Ye Xu
{"title":"De novo familial adenomatous polyposis with germline double heterozygosity of APC/BRCA2: a case report and literature review.","authors":"Tian-Qi Zhang, Ji-Dong Cai, Cong Li, Yun Xu, Ye Xu","doi":"10.1186/s13053-025-00306-x","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The widespread application of colonoscopy screening and genetic testing in colorectal cancer (CRC) treatment has led to the identification of a subset of familial adenomatous polyposis (FAP) patients who lack a family history of the disease but harbor germline gene mutations. Moreover, distinct genotypes may be associated with varied clinical presentations and therapeutic options. This case report describes a male patient with de novo FAP who harbored germline double heterozygosity (GDH) for APC and BRCA2 mutations. The patient underwent total colectomy, and genetic testing enabled personalized surveillance and management strategies for his family members.</p><p><strong>Case presentation: </strong>A 43-year-old male with no family history of cancer presented to the outpatient clinic of the Colorectal Surgery Department with complaints of constipation and hematochezia. Colonoscopy revealed hundreds of polyps throughout the colon and a rectal adenocarcinoma located 5 cm from the anal verge. Gastroduodenal endoscopy did not detect any upper gastrointestinal adenomas. The patient underwent laparoscopic total colectomy with abdominoperineal resection of the rectum and end ileostomy. With the consent of the patient and his family, genetic testing was performed. The index patient was found to carry an APC splicing site mutation (exon 15: c.1744-1G > A) and a BRCA2 missense mutation (exon 17: c.7976G > A: p.R2659K). His daughter was found to have inherited the same germline BRCA2 variant. Additionally, the rectal cancer exhibited proficient DNA mismatch repair (pMMR) status, ERBB2 copy number amplification, and a missense mutation, while the KRAS, NRAS, and BRAF genes were wild-type. Based on the genetic testing results and clinical manifestations, the index patient was diagnosed with familial adenomatous polyposis (FAP) and rectal cancer. Personalized surveillance and management strategies were implemented for the patient and his family, focusing on the risks of extra-colonic diseases and potential malignancies in the prostate, pancreas, breast, and ovaries.</p><p><strong>Conclusion: </strong>De novo FAP with double germline mutations in APC and BRCA2, along with somatic ERBB2 mutations, is exceptionally rare among hereditary cancer cases. With the rapid advancements in genomics, the detection of multiple gene variants in individuals or families has become increasingly common. Additionally, the application of artificial intelligence (AI) in medical research may provide powerful tools for genetic analysis and clinical decision-making. Consequently, a comprehensive evaluation of family history, a deep understanding of hereditary cancer syndromes, and precise interpretation of genetic mutations are essential for personalized clinical management in the era of precision medicine. However, these tasks pose significant challenges for clinicians and genetic counselors alike.</p>","PeriodicalId":55058,"journal":{"name":"Hereditary Cancer in Clinical Practice","volume":"23 1","pages":"6"},"PeriodicalIF":2.4000,"publicationDate":"2025-02-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11843810/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Hereditary Cancer in Clinical Practice","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13053-025-00306-x","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"ONCOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: The widespread application of colonoscopy screening and genetic testing in colorectal cancer (CRC) treatment has led to the identification of a subset of familial adenomatous polyposis (FAP) patients who lack a family history of the disease but harbor germline gene mutations. Moreover, distinct genotypes may be associated with varied clinical presentations and therapeutic options. This case report describes a male patient with de novo FAP who harbored germline double heterozygosity (GDH) for APC and BRCA2 mutations. The patient underwent total colectomy, and genetic testing enabled personalized surveillance and management strategies for his family members.
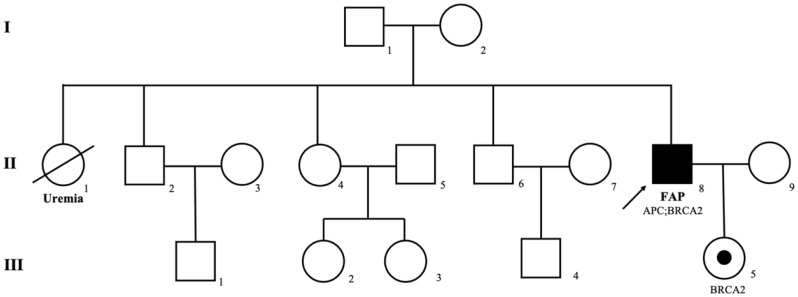
Case presentation: A 43-year-old male with no family history of cancer presented to the outpatient clinic of the Colorectal Surgery Department with complaints of constipation and hematochezia. Colonoscopy revealed hundreds of polyps throughout the colon and a rectal adenocarcinoma located 5 cm from the anal verge. Gastroduodenal endoscopy did not detect any upper gastrointestinal adenomas. The patient underwent laparoscopic total colectomy with abdominoperineal resection of the rectum and end ileostomy. With the consent of the patient and his family, genetic testing was performed. The index patient was found to carry an APC splicing site mutation (exon 15: c.1744-1G > A) and a BRCA2 missense mutation (exon 17: c.7976G > A: p.R2659K). His daughter was found to have inherited the same germline BRCA2 variant. Additionally, the rectal cancer exhibited proficient DNA mismatch repair (pMMR) status, ERBB2 copy number amplification, and a missense mutation, while the KRAS, NRAS, and BRAF genes were wild-type. Based on the genetic testing results and clinical manifestations, the index patient was diagnosed with familial adenomatous polyposis (FAP) and rectal cancer. Personalized surveillance and management strategies were implemented for the patient and his family, focusing on the risks of extra-colonic diseases and potential malignancies in the prostate, pancreas, breast, and ovaries.
Conclusion: De novo FAP with double germline mutations in APC and BRCA2, along with somatic ERBB2 mutations, is exceptionally rare among hereditary cancer cases. With the rapid advancements in genomics, the detection of multiple gene variants in individuals or families has become increasingly common. Additionally, the application of artificial intelligence (AI) in medical research may provide powerful tools for genetic analysis and clinical decision-making. Consequently, a comprehensive evaluation of family history, a deep understanding of hereditary cancer syndromes, and precise interpretation of genetic mutations are essential for personalized clinical management in the era of precision medicine. However, these tasks pose significant challenges for clinicians and genetic counselors alike.
期刊介绍:
Hereditary Cancer in Clinical Practice is an open access journal that publishes articles of interest for the cancer genetics community and serves as a discussion forum for the development appropriate healthcare strategies.
Cancer genetics encompasses a wide variety of disciplines and knowledge in the field is rapidly growing, especially as the amount of information linking genetic differences to inherited cancer predispositions continues expanding. With the increased knowledge of genetic variability and how this relates to cancer risk there is a growing demand not only to disseminate this information into clinical practice but also to enable competent debate concerning how such information is managed and what it implies for patient care.
Topics covered by the journal include but are not limited to:
Original research articles on any aspect of inherited predispositions to cancer.
Reviews of inherited cancer predispositions.
Application of molecular and cytogenetic analysis to clinical decision making.
Clinical aspects of the management of hereditary cancers.
Genetic counselling issues associated with cancer genetics.
The role of registries in improving health care of patients with an inherited predisposition to cancer.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


