Electronic and Optical Properties of Functionalized Dihydroxyanthraquinone as Potential Organic Semiconductor Materials for Solar Cells Applications
IF 3.3
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
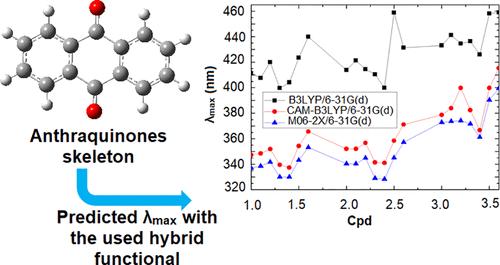
The electronic and optical properties of three dihydroxyanthraquinone organic molecules─anthrarufin, chrysazin, and quinizarin─were theoretically investigated as potential candidates for organic solar cells. Using density functional theory (DFT), key electronic properties such as reorganization energy (λ), adiabatic ionization potential (IA), adiabatic electron affinity (EA), HOMO, LUMO, and energy gap (Egap) were calculated. Time-dependent DFT (TD-DFT) was employed to determine optical properties, including maximum absorption (λmax) and oscillator strengths (f) at excited states in both a vacuum and solvent. Additionally, nonlinear optical properties, such as polarizability and first hyperpolarizability, were evaluated. Global reactivity descriptors, including dipole moment (μ), electronegativity (χ), hardness (η), softness (S), and electrophilicity index (ω), were also calculated. The impact of functionalization on these molecules’ properties was examined to understand its effects and provide guidelines for designing and synthesizing new derivatives with enhanced properties for use as semiconductors in organic electronics. The results indicate a correlation between the type and position of functional groups, reorganization energies, and predicted charge transport properties. Functional groups such as CN lower both λh and λe, making them promising candidates for n-type materials. The position and type of functional groups significantly affect global properties like dipole moment and energy gap (Egap), with quinizarin exhibiting the lowest dipole moment (0.97 D) and variations reaching up to 10.76 D. To further understand the intermolecular interactions of the examined molecules, Hirshfeld surface analysis and energy framework studies were conducted.
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


