Optimizing cation–π force fields for molecular dynamics studies of competitive solvation in conjugated organosulfur polymers for lithium–sulfur batteries†
Diptesh Gayen, Yannik Schütze, Sébastien Groh and Joachim Dzubiella
{"title":"Optimizing cation–π force fields for molecular dynamics studies of competitive solvation in conjugated organosulfur polymers for lithium–sulfur batteries†","authors":"Diptesh Gayen, Yannik Schütze, Sébastien Groh and Joachim Dzubiella","doi":"10.1039/D4CP04484C","DOIUrl":null,"url":null,"abstract":"<p >Lithium–sulfur (Li/S) batteries are emerging as a next-generation energy storage technology due to their high theoretical energy density and cost-effectiveness. π-Conjugated organosulfur polymers, such as poly(4-(thiophene-3-yl)benzenethiol) (PTBT), have shown promise in overcoming challenges such as the polysulfide shuttle effect by providing a conductive framework and enabling sulfur copolymerization. In these cathodes, cation–π interactions significantly influence Li<small><sup>+</sup></small> diffusion and storage properties in π-conjugated cathodes, but classical OPLS-AA force fields fail to capture these effects. This study employs a bottom-up approach based on density functional theory (DFT) to optimize the nonbonded interaction parameters (OPLS-AA/corr.), particularly for the Li<small><sup>+</sup></small>–π interactions with the PTBT polymer. Following prior work, we used an ion-induced dipole potential to model the cation–π interactions. The impact of the solvent on the PTBT monomers was examined by computing the potential of mean force (PMF) between PTBT monomers and Li<small><sup>+</sup></small> ions in both explicit and implicit solvents using the Boltzmann inversion of probability distributions close to room temperature. In the implicit solvent case, the magnitude of the binding free energy decreased with increasing dielectric constant, as the dominant electrostatics scaled with the dielectric constant. In contrast, in the explicit solvent case, considering the mixtures of organic solvent DME and DOL, the binding free energy shows minimal dependence on solvent composition due to the competing interaction of TBT and Li<small><sup>+</sup></small> with the solvent molecules. However, increasing salt concentration decreases the binding free energy due to Debye–Hückel screening effects. In general, this work suggests that the optimized parameters can be widely used in the simulation of polymers in electrolytes for the Li/S battery to enhance the representation of cation–π interactions for a fixed charge force field.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 11","pages":" 5655-5668"},"PeriodicalIF":2.9000,"publicationDate":"2025-02-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d4cp04484c?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp04484c","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
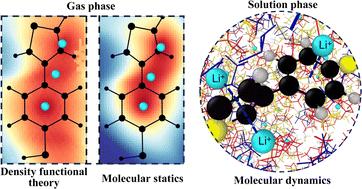
Lithium–sulfur (Li/S) batteries are emerging as a next-generation energy storage technology due to their high theoretical energy density and cost-effectiveness. π-Conjugated organosulfur polymers, such as poly(4-(thiophene-3-yl)benzenethiol) (PTBT), have shown promise in overcoming challenges such as the polysulfide shuttle effect by providing a conductive framework and enabling sulfur copolymerization. In these cathodes, cation–π interactions significantly influence Li+ diffusion and storage properties in π-conjugated cathodes, but classical OPLS-AA force fields fail to capture these effects. This study employs a bottom-up approach based on density functional theory (DFT) to optimize the nonbonded interaction parameters (OPLS-AA/corr.), particularly for the Li+–π interactions with the PTBT polymer. Following prior work, we used an ion-induced dipole potential to model the cation–π interactions. The impact of the solvent on the PTBT monomers was examined by computing the potential of mean force (PMF) between PTBT monomers and Li+ ions in both explicit and implicit solvents using the Boltzmann inversion of probability distributions close to room temperature. In the implicit solvent case, the magnitude of the binding free energy decreased with increasing dielectric constant, as the dominant electrostatics scaled with the dielectric constant. In contrast, in the explicit solvent case, considering the mixtures of organic solvent DME and DOL, the binding free energy shows minimal dependence on solvent composition due to the competing interaction of TBT and Li+ with the solvent molecules. However, increasing salt concentration decreases the binding free energy due to Debye–Hückel screening effects. In general, this work suggests that the optimized parameters can be widely used in the simulation of polymers in electrolytes for the Li/S battery to enhance the representation of cation–π interactions for a fixed charge force field.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


