Case report: Novel ACTN4 variant of uncertain significance in a pediatric case of steroid-resistant nephrotic syndrome requesting kidney transplantation.
Ignacio Alarcón, Carolina Peralta, Francisco Cammarata-Scalisi, Maykol Araya Castillo, Francisco Cano, Angélica Rojo, María Luisa Ceballos, Paola Krall
{"title":"Case report: Novel <i>ACTN4</i> variant of uncertain significance in a pediatric case of steroid-resistant nephrotic syndrome requesting kidney transplantation.","authors":"Ignacio Alarcón, Carolina Peralta, Francisco Cammarata-Scalisi, Maykol Araya Castillo, Francisco Cano, Angélica Rojo, María Luisa Ceballos, Paola Krall","doi":"10.3389/fneph.2024.1375538","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Steroid-resistant nephrotic syndrome (SRNS) is a rare kidney disease commonly characterized histopathologically by focal and segmental glomerulosclerosis (FSGS) or minimal change disease. One-third of SRNS-FSGS cases are attributed to a genetic cause ultimately leading to end-stage kidney disease (ESKD) during childhood or adulthood. <i>ACTN4</i> variants, although rare, typically manifest in early adulthood as SRNS-FSGS with autosomal dominant inheritance pattern and are associated with variable progression toward ESKD.</p><p><strong>Case–diagnosis/treatment: </strong>A 10-year-old Chilean male patient, born to a complicated pregnancy without any history of prenatal care, was incidentally found to have mild proteinuria during pre-surgery analysis. He was diagnosed with nephrotic syndrome and treatment with prednisone was started, but 12 months later, he persisted with hyperlipidemia, hypoalbuminemia, and proteinuria. Within a few weeks, proteinuria rapidly increased, and a kidney biopsy exhibited FSGS features. At the age of 12, he reached ESKD and initiated peritoneal dialysis, experiencing an episode of posterior reversible encephalopathy syndrome. Exome sequencing identified a novel variant of uncertain significance (VUS), <i>ACTN4</i> c.625_633del that predicted the in-frame deletion p.L209_E211del in a highly conserved functional domain. He requested to be considered for kidney transplantation and the VUS in <i>ACTN4</i> was re-analyzed to assess potential risks, resulting in a reclassification as likely pathogenic (PM1+PM2+PM4 criteria). At 14 years old, he received a deceased donor kidney allograft without recurrence during the subsequent 5 months.</p><p><strong>Conclusions: </strong>Identifying VUS is a recurring challenge in routine clinical genetics, particularly for patients with rare diseases or atypical phenotypes in underrepresented populations. This case underscores the benefit of timely genetic diagnosis taking into account the patient's request. VUS reassessment becomes more relevant when considering a kidney transplant not only as an appropriate procedure, but as the therapy of choice, especially considering the patient's history of complications with variable long-term consequences.</p>","PeriodicalId":73091,"journal":{"name":"Frontiers in nephrology","volume":"4 ","pages":"1375538"},"PeriodicalIF":0.0000,"publicationDate":"2025-01-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11826236/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in nephrology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/fneph.2024.1375538","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Steroid-resistant nephrotic syndrome (SRNS) is a rare kidney disease commonly characterized histopathologically by focal and segmental glomerulosclerosis (FSGS) or minimal change disease. One-third of SRNS-FSGS cases are attributed to a genetic cause ultimately leading to end-stage kidney disease (ESKD) during childhood or adulthood. ACTN4 variants, although rare, typically manifest in early adulthood as SRNS-FSGS with autosomal dominant inheritance pattern and are associated with variable progression toward ESKD.
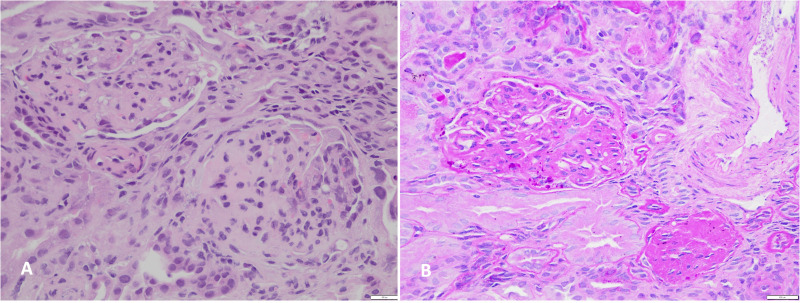
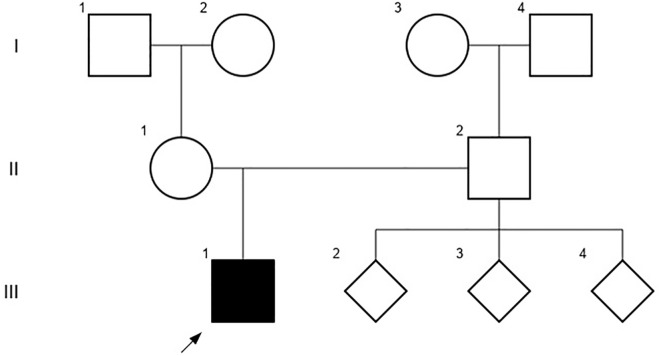
Case–diagnosis/treatment: A 10-year-old Chilean male patient, born to a complicated pregnancy without any history of prenatal care, was incidentally found to have mild proteinuria during pre-surgery analysis. He was diagnosed with nephrotic syndrome and treatment with prednisone was started, but 12 months later, he persisted with hyperlipidemia, hypoalbuminemia, and proteinuria. Within a few weeks, proteinuria rapidly increased, and a kidney biopsy exhibited FSGS features. At the age of 12, he reached ESKD and initiated peritoneal dialysis, experiencing an episode of posterior reversible encephalopathy syndrome. Exome sequencing identified a novel variant of uncertain significance (VUS), ACTN4 c.625_633del that predicted the in-frame deletion p.L209_E211del in a highly conserved functional domain. He requested to be considered for kidney transplantation and the VUS in ACTN4 was re-analyzed to assess potential risks, resulting in a reclassification as likely pathogenic (PM1+PM2+PM4 criteria). At 14 years old, he received a deceased donor kidney allograft without recurrence during the subsequent 5 months.
Conclusions: Identifying VUS is a recurring challenge in routine clinical genetics, particularly for patients with rare diseases or atypical phenotypes in underrepresented populations. This case underscores the benefit of timely genetic diagnosis taking into account the patient's request. VUS reassessment becomes more relevant when considering a kidney transplant not only as an appropriate procedure, but as the therapy of choice, especially considering the patient's history of complications with variable long-term consequences.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


