Andrea Cortese, Maike F Dohrn, Riccardo Curro, Sara Negri, Petra Lassuthova, Chiara Pisciotta, Stefano Tozza, Abdullah Al-Ajmi, Changyong Feng, Pedro J Tomaselli, Gorka Fernandez-Eulate, Saif Haddad, Matilde Laurà, Alexander M Rossor, Elisa Vegezzi, Stefano Facchini, James N Sleigh, Adriana Rebelo, Danique Beijer, Jacquelyn Raposo, Mario Saporta, Barbora Lauerova, Helena F Pernice, Pascal Achenbach, Ulrike Schöne, Tayir Alon, Marcus Deschauer, Isabell Cordts, Carolin D Obermaier, Natalie Winter, Peter D Creigh, Janet E Sowden, Tyler Rehbein, Stefania Magri, Alessandro Bertini, Paola Saveri, Paolo Ripellino, Jingyu Huang, Aleksandra Nadaj-Pakleza, Alison Ross, James K L Holt, Kathryn M Brennan, Rivka Sukenik-Halevy, Varoona Bizaoui, Yesim Parman, Esra Battaloglu, Arman Cakar, Hadil Alrohaif, Simon Hammans, Kishore R Kumar, Marina L Kennerson, Hülya Kayserili, Defne A Amado, Katrin Hahn, Paola Valentino, Francesca Cavalcanti, Carlo Gaetano, Franco Taroni, Geir J Braathen, Henry Houlden, Tanya Stojkovic, Stojan Peric, Alessandra Bolino, Stefano C Previtali, Lee Yi-Chung, Ayşe N Başak, Sherifa A Hamed, Ricardo Rojas-Garcia, Kristl G Claeys, Wilson Marques, Teresa Sevilla, Beate Schlotter-Weigel, Fiore Manganelli, Ruxu Zhang, David N Herrmann, Steven S Scherer, Pavel Seeman, Davide Pareyson, Mary M Reilly, Michael E Shy, Stephan Züchner
{"title":"Genotype and phenotype spectrum of Charcot-Marie-Tooth disease due to mutations in SORD.","authors":"Andrea Cortese, Maike F Dohrn, Riccardo Curro, Sara Negri, Petra Lassuthova, Chiara Pisciotta, Stefano Tozza, Abdullah Al-Ajmi, Changyong Feng, Pedro J Tomaselli, Gorka Fernandez-Eulate, Saif Haddad, Matilde Laurà, Alexander M Rossor, Elisa Vegezzi, Stefano Facchini, James N Sleigh, Adriana Rebelo, Danique Beijer, Jacquelyn Raposo, Mario Saporta, Barbora Lauerova, Helena F Pernice, Pascal Achenbach, Ulrike Schöne, Tayir Alon, Marcus Deschauer, Isabell Cordts, Carolin D Obermaier, Natalie Winter, Peter D Creigh, Janet E Sowden, Tyler Rehbein, Stefania Magri, Alessandro Bertini, Paola Saveri, Paolo Ripellino, Jingyu Huang, Aleksandra Nadaj-Pakleza, Alison Ross, James K L Holt, Kathryn M Brennan, Rivka Sukenik-Halevy, Varoona Bizaoui, Yesim Parman, Esra Battaloglu, Arman Cakar, Hadil Alrohaif, Simon Hammans, Kishore R Kumar, Marina L Kennerson, Hülya Kayserili, Defne A Amado, Katrin Hahn, Paola Valentino, Francesca Cavalcanti, Carlo Gaetano, Franco Taroni, Geir J Braathen, Henry Houlden, Tanya Stojkovic, Stojan Peric, Alessandra Bolino, Stefano C Previtali, Lee Yi-Chung, Ayşe N Başak, Sherifa A Hamed, Ricardo Rojas-Garcia, Kristl G Claeys, Wilson Marques, Teresa Sevilla, Beate Schlotter-Weigel, Fiore Manganelli, Ruxu Zhang, David N Herrmann, Steven S Scherer, Pavel Seeman, Davide Pareyson, Mary M Reilly, Michael E Shy, Stephan Züchner","doi":"10.1093/brain/awaf021","DOIUrl":null,"url":null,"abstract":"<p><p>Biallelic loss-of-function mutations in the sorbitol dehydrogenase (SORD) gene cause the most common recessive type of Charcot-Marie-Tooth disease (CMT), CMT-SORD. However, the full genotype-phenotype spectrum and progression of the disease remain to be defined. Notably, a multicentre phase 2/3 study to test the efficacy of govorestat (NCT05397665), a new aldose reductase inhibitor, is currently ongoing. Diagnosing CMT-SORD will become imperative when disease-modifying therapies become available. In this cross-sectional multicentre study, we identified 144 patients from 126 families, including 99 males (69%) and 45 females (31%). Patients represented multiple ancestries, including European, Hispanic, Chinese, Near Eastern and Northern African. We confirmed c.757delG (p.Ala253GlnfsTer27) as the most common pathogenic allele, followed by c.458C>A (p.Ala153Asp), while other variants were identified, mostly in single cases. The average sorbitol level in CMT-SORD patients was significantly higher compared to controls and heterozygous carriers, independently from serum storage duration, sex or variant type. Two-thirds of cases were diagnosed with CMT2 while one-third had distal hereditary motor neuropathy. Disease onset was usually in the second decade of life. Although foot dorsiflexion was the most affected muscle group, dorsal and plantar flexion had a similar degree of weakness in most cases (difference of Medical Research Council score ≤ 1). One-fourth of patients used ankle foot orthoses, usually in their 30s, but most patients maintained independent ambulation later in life. Nerve conduction studies were suggestive of a motor predominant axonal neuropathy, with reduced conduction velocities in the intermediate range in a quarter of the cases. Sensory conductions in the upper limbs appeared more frequently affected than in the lower limbs. Foot dorsiflexion and plantar flexion decreased significantly with age. Male sex was significantly associated with the severity of distal lower limb weakness (plantar flexion) and a larger change over time (dorsiflexion). In conclusion, CMT-SORD is a frequent recessive form of axonal, motor predominant CMT, with prominent foot dorsiflexion and plantar flexion involvement. Fasting serum sorbitol is a reliable biomarker of the condition that can be utilized for pathogenicity assessment of identified rare SORD variants.</p>","PeriodicalId":9063,"journal":{"name":"Brain","volume":" ","pages":"3737-3747"},"PeriodicalIF":11.7000,"publicationDate":"2025-10-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12493047/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Brain","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1093/brain/awaf021","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
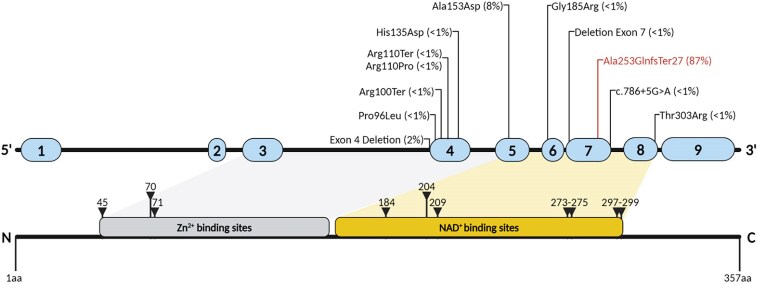
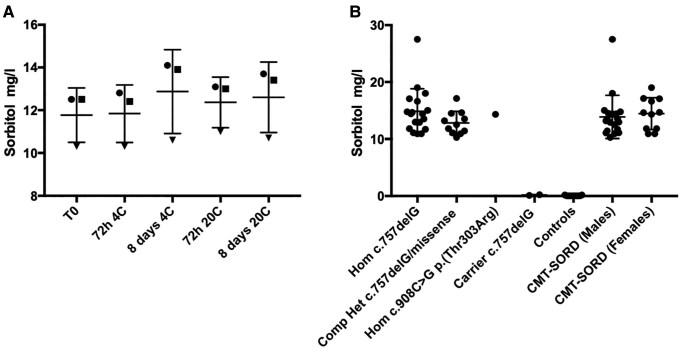
Biallelic loss-of-function mutations in the sorbitol dehydrogenase (SORD) gene cause the most common recessive type of Charcot-Marie-Tooth disease (CMT), CMT-SORD. However, the full genotype-phenotype spectrum and progression of the disease remain to be defined. Notably, a multicentre phase 2/3 study to test the efficacy of govorestat (NCT05397665), a new aldose reductase inhibitor, is currently ongoing. Diagnosing CMT-SORD will become imperative when disease-modifying therapies become available. In this cross-sectional multicentre study, we identified 144 patients from 126 families, including 99 males (69%) and 45 females (31%). Patients represented multiple ancestries, including European, Hispanic, Chinese, Near Eastern and Northern African. We confirmed c.757delG (p.Ala253GlnfsTer27) as the most common pathogenic allele, followed by c.458C>A (p.Ala153Asp), while other variants were identified, mostly in single cases. The average sorbitol level in CMT-SORD patients was significantly higher compared to controls and heterozygous carriers, independently from serum storage duration, sex or variant type. Two-thirds of cases were diagnosed with CMT2 while one-third had distal hereditary motor neuropathy. Disease onset was usually in the second decade of life. Although foot dorsiflexion was the most affected muscle group, dorsal and plantar flexion had a similar degree of weakness in most cases (difference of Medical Research Council score ≤ 1). One-fourth of patients used ankle foot orthoses, usually in their 30s, but most patients maintained independent ambulation later in life. Nerve conduction studies were suggestive of a motor predominant axonal neuropathy, with reduced conduction velocities in the intermediate range in a quarter of the cases. Sensory conductions in the upper limbs appeared more frequently affected than in the lower limbs. Foot dorsiflexion and plantar flexion decreased significantly with age. Male sex was significantly associated with the severity of distal lower limb weakness (plantar flexion) and a larger change over time (dorsiflexion). In conclusion, CMT-SORD is a frequent recessive form of axonal, motor predominant CMT, with prominent foot dorsiflexion and plantar flexion involvement. Fasting serum sorbitol is a reliable biomarker of the condition that can be utilized for pathogenicity assessment of identified rare SORD variants.
期刊介绍:
Brain, a journal focused on clinical neurology and translational neuroscience, has been publishing landmark papers since 1878. The journal aims to expand its scope by including studies that shed light on disease mechanisms and conducting innovative clinical trials for brain disorders. With a wide range of topics covered, the Editorial Board represents the international readership and diverse coverage of the journal. Accepted articles are promptly posted online, typically within a few weeks of acceptance. As of 2022, Brain holds an impressive impact factor of 14.5, according to the Journal Citation Reports.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


