Astrid Høj, Sonja Holm-Yildiz, Thomas Krag, Danijela Dejanovic, Thomas van Overeem Hansen, Morten Dunø, Mette Cathrine Ørngreen, John Vissing, Nicoline Løkken
{"title":"2-[18F] FDG PET/CT in Rapid Late-Onset Multiple Acyl-CoA Dehydrogenase Deficiency: A Case Report","authors":"Astrid Høj, Sonja Holm-Yildiz, Thomas Krag, Danijela Dejanovic, Thomas van Overeem Hansen, Morten Dunø, Mette Cathrine Ørngreen, John Vissing, Nicoline Løkken","doi":"10.1002/jmd2.12469","DOIUrl":null,"url":null,"abstract":"<p>Multiple acyl-CoA dehydrogenase deficiency (MADD) is a rare inborn metabolic myopathy affecting fat and protein metabolism. Patients with late-onset MADD typically present with exercise intolerance and muscle weakness. We present a patient with an acute, very late-onset symptom debut at 52 years of age. Over 5 months, the patient deteriorated from asymptomatic to almost complete loss of ambulation. He had a substantial weight loss, head-drop, progressive proximal limb and chewing weakness. Due to the rapid progression, amyotrophic lateral sclerosis, myositis, myasthenia gravis and a paraneoplastic syndrome in relation to underlying malignancy were considered first. A 2-[<sup>18</sup>F] FDG PET/CT scan was performed to exclude a paraneoplastic syndrome. The scan revealed diffuse and symmetric, pathologically high 2-[<sup>18</sup>F] FDG-uptake in the patient's neck, shoulder, and paravertebral muscles, which was later suggested as a sign of a metabolic myopathy. Muscle biopsy (Oil Red O staining) and acylcarnitine profile (elevated C5-C18 acylcarnitines) findings suggested MADD, which was confirmed by genetic analysis showing biallelic variants in the <i>ETFDH</i> gene (c.1763A>G, p.(His588Arg); c.897G>A, p.(Leu299=)). After 1 month of dietary intervention and daily diet supplements (riboflavin 400 mg TID, levocarnitine 1 g TID, Q10 150 mg qD in two doses), the patient had almost recovered to his habitual level. A posttreatment muscle biopsy showed less disrupted ultrastructure of the myofibers. We learned from this case of rapid and late-onset MADD that 2-[<sup>18</sup>F] FDG PET/CT, with diffuse and symmetric 2-[<sup>18</sup>F] FDG-uptake in skeletal muscle, can be valuable in clarifying this rare diagnosis.</p>","PeriodicalId":14930,"journal":{"name":"JIMD reports","volume":"66 2","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2025-02-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jmd2.12469","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JIMD reports","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jmd2.12469","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 0
Abstract
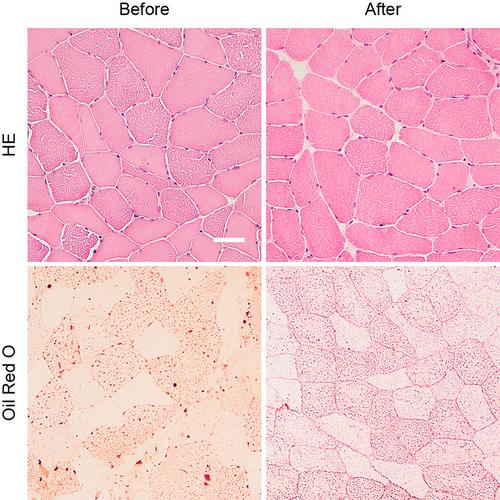
Multiple acyl-CoA dehydrogenase deficiency (MADD) is a rare inborn metabolic myopathy affecting fat and protein metabolism. Patients with late-onset MADD typically present with exercise intolerance and muscle weakness. We present a patient with an acute, very late-onset symptom debut at 52 years of age. Over 5 months, the patient deteriorated from asymptomatic to almost complete loss of ambulation. He had a substantial weight loss, head-drop, progressive proximal limb and chewing weakness. Due to the rapid progression, amyotrophic lateral sclerosis, myositis, myasthenia gravis and a paraneoplastic syndrome in relation to underlying malignancy were considered first. A 2-[18F] FDG PET/CT scan was performed to exclude a paraneoplastic syndrome. The scan revealed diffuse and symmetric, pathologically high 2-[18F] FDG-uptake in the patient's neck, shoulder, and paravertebral muscles, which was later suggested as a sign of a metabolic myopathy. Muscle biopsy (Oil Red O staining) and acylcarnitine profile (elevated C5-C18 acylcarnitines) findings suggested MADD, which was confirmed by genetic analysis showing biallelic variants in the ETFDH gene (c.1763A>G, p.(His588Arg); c.897G>A, p.(Leu299=)). After 1 month of dietary intervention and daily diet supplements (riboflavin 400 mg TID, levocarnitine 1 g TID, Q10 150 mg qD in two doses), the patient had almost recovered to his habitual level. A posttreatment muscle biopsy showed less disrupted ultrastructure of the myofibers. We learned from this case of rapid and late-onset MADD that 2-[18F] FDG PET/CT, with diffuse and symmetric 2-[18F] FDG-uptake in skeletal muscle, can be valuable in clarifying this rare diagnosis.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


