Role of explicit solvation and level of theory in predicting the aqueous reduction potential of carbonate radical anion by DFT
IF 2.9
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Chemical oxidation reactions, a key class of electron transfer processes, have broad applications, including the treatment of persistent and mobile pollutants. Marcus theory, paired with density functional theory (DFT) simulations, enables quantification of thermodynamic properties in these reactions. However, accurately modeling species with complex solvent interactions, especially radicals, requires careful selection of computational methods. Reduction potentials provide critical benchmarks for evaluating solvent models and functional choices by comparing simulated values to literature data. In this study, we used the carbonate radical, known for its strong intermolecular interactions, as a model to assess solvation models and computational functionals. Implicit solvation methods significantly underperformed, predicting only one-third of the measured reduction potential. Accurate results were obtained using explicit solvation with 18 water molecules for ωB97xD/6-311++G(2d,2p) and 9 water molecules for M06-2X/6-311++G(2d,2p). B3LYP/6-311++G(2d,2p) showed improvement with additional explicit solvation but failed to match literature benchmarks. Functional performance differences, analyzed through natural bond orbital (NBO) and charge transfer calculations, emphasized the critical role of dispersion corrections. Testing various dispersion correction methods revealed consistent improvements in reduction potential accuracy. These findings highlight the necessity of explicit solvation for modeling electron transfer reactions with extensive solvent interactions and underscore the importance of selecting appropriate functionals and dispersion corrections for reliable predictions.
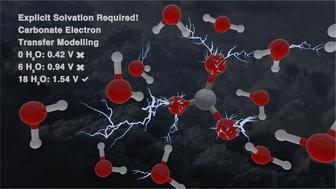
化学氧化反应是一类关键的电子转移过程,应用广泛,包括处理持久性和流动性污染物。马库斯理论与密度泛函理论(DFT)模拟相结合,可以量化这些反应的热力学性质。然而,要准确模拟具有复杂溶剂相互作用的物种,尤其是自由基,需要谨慎选择计算方法。通过比较模拟值和文献数据,还原电势为评估溶剂模型和函数选择提供了关键基准。在本研究中,我们以分子间相互作用强烈的碳酸根为模型,对溶解模型和计算函数进行了评估。隐式溶解方法表现明显不佳,只能预测出测量还原电位的三分之一。在ωB97xD/6-311++G(2d,2p)和 M06-2X/6-311++G(2d,2p)中,分别使用了 18 个水分子和 9 个水分子的显式溶胶法获得了精确的结果。B3LYP/6-311++G(2d,2p) 通过额外的显式溶解得到了改进,但未能达到文献基准。通过天然键轨道(NBO)和电荷转移计算分析的函数性能差异强调了色散修正的关键作用。对各种分散修正方法的测试表明,还原电势的准确性得到了持续改善。这些发现凸显了显式溶解对具有广泛溶剂相互作用的电子转移反应建模的必要性,并强调了选择适当的函数和色散修正进行可靠预测的重要性。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Physical Chemistry Chemical Physics
化学-物理:原子、分子和化学物理
CiteScore
5.50
自引率
9.10%
发文量
2675
审稿时长
2.0 months
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


