Density Functional Theory Study of Surface Stability and Phase Diagram of Orthorhombic CsPbI3
IF 3.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
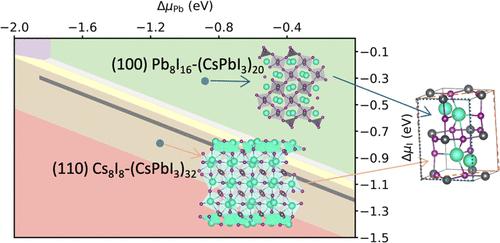
CsPbI3 has been recognized as a promising candidate for optoelectronic device applications. To further enhance device efficiency, it is imperative to understand the surface properties of CsPbI3, which affect the charge transport and defect formation. In this study, we conducted density functional theory calculations to explore the stability of the (001), (110), and (100) surfaces of orthorhombic CsPbI3, considering different stoichiometries and surface reconstructions. Our results show that under the chemical potentials confined by the thermodynamically stable region of bulk CsPbI3, the CsI-terminated surfaces of (001) and (110) and the stoichiometric surface of (100) are stable. Among these three surfaces, the CsI-terminated (110) surface has the lowest surface energy and no midgap states, which benefits the transport properties of the material.
正交CsPbI3表面稳定性和相图的密度泛函理论研究
CsPbI3已被公认为光电子器件应用的有前途的候选者。为了进一步提高器件效率,有必要了解CsPbI3的表面性质,它会影响电荷输运和缺陷形成。在本研究中,我们通过密度泛函理论计算来探索正交CsPbI3(001)、(110)和(100)表面的稳定性,考虑不同的化学计量和表面重建。结果表明,在CsPbI3体的热力学稳定区所限定的化学势下,(001)和(110)的csi端接面以及(100)的化学计量面是稳定的。在这三种表面中,端接csi(110)的表面具有最低的表面能,并且没有中间间隙状态,这有利于材料的输运性质。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


