Natural history of inflammation and impaired autophagy in children with Gaucher disease identified by newborn screening
IF 1.9
4区 医学
Q3 GENETICS & HEREDITY
引用次数: 0
Abstract
Introduction
Gaucher disease is a lysosomal storage disease due to deficiency of glucocerebrosidase, leading to the accumulation of glucosylceramide, particularly in macrophages. In addition to storage, secondary abnormalities such as inflammation, cellular stress, and impaired autophagy may contribute to the disease pathogenesis. The onset and course of progression of these secondary abnormalities remains unclear. Owing to the increasingly widespread newborn screening programs, diagnosis can be made at a presymptomatic stage. Understanding the early natural course of the disease is important for optimal monitoring and management of such at-risk individuals.
The aim of our study is to investigate secondary abnormalities in very young children with type 1 Gaucher disease identified through neonatal screening.
Materials and methods
We enrolled five children (<4 years old) with type I Gaucher disease in a presymptomatic stage and not receiving therapy. We assessed plasma cytokine profiles (TNFα, IL1β, and IL6 by ELISA), activation of pro-inflammatory p38 mitogen-activated protein kinase (MAPK) and the abundance of LC3-II as indicator of autophagic flux, by immunoblotting.
Results
All subjects exhibited elevated TNFα (mean 21.74 μmol/L, SD 37.48, range 2.37–88.72 μmol/L). The other cytokines analyzed were within normal range. Cellular stress (activation of p38) was present in the child with higher glucosylsphingosine (GluSph) accumulation. Additionally, all subjects showed a significant reduction in LC3-II (mean 88 %, SD 9 %, range 77–98 %), indicating reduced autophagic flux.
Discussion
We have identified the presence of inflammation with inhibition of autophagic flux in presymptomatic young children with a genetically confirmed high-risk of developing Gaucher disease. These findings contribute insights into the early course of Gaucher disease and support the management of at-risk individuals identified by newborn screening. Therapeutic interventions including specific enzyme replacement or other means to address inflammation or autophagy could delay or prevent the onset of symptomatic disease and consequential disability. Further clinical studies are warranted to explore these possibilities.
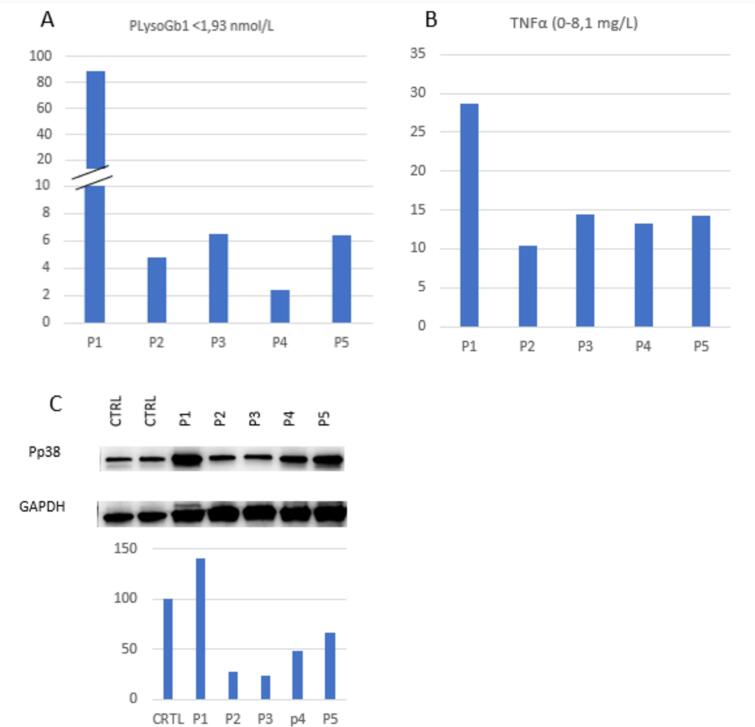
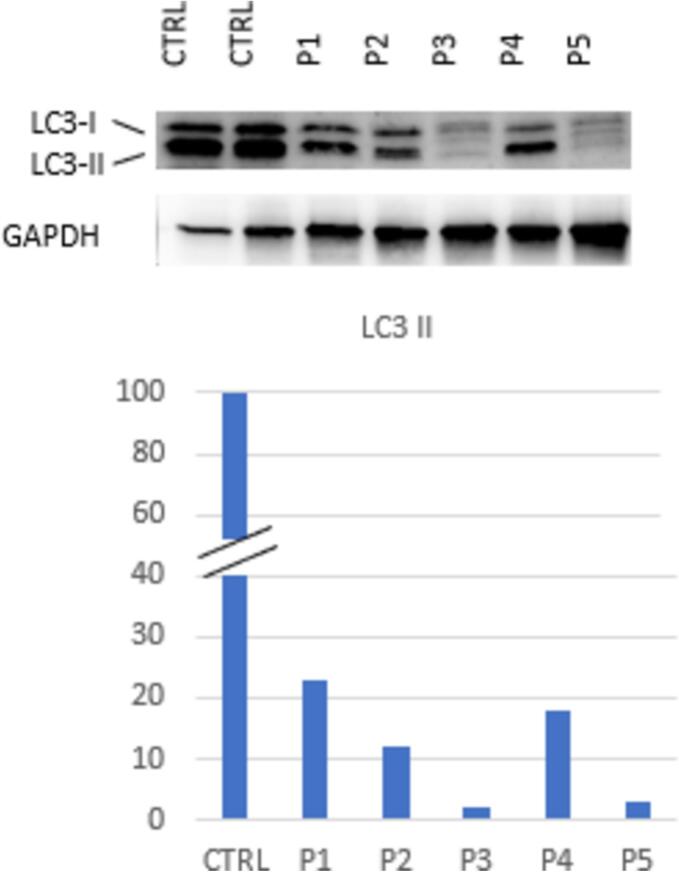
新生儿筛查确定的戈谢病患儿炎症和自噬受损的自然史
戈谢病是一种溶酶体贮积病,由于葡萄糖脑苷酶缺乏,导致葡萄糖神经酰胺积聚,特别是在巨噬细胞中。除了储存外,继发性异常,如炎症、细胞应激和自噬受损可能有助于疾病的发病机制。这些继发性异常的发病和发展过程尚不清楚。由于新生儿筛查项目日益广泛,可以在症状前阶段进行诊断。了解疾病的早期自然过程对于这些高危个体的最佳监测和管理非常重要。我们研究的目的是研究通过新生儿筛查发现的非常年幼的1型戈谢病儿童的继发性异常。材料和方法:5名儿童入组。结果:所有受试者均出现TNFα升高(平均21.74 μmol/L, SD 37.48,范围2.37 ~ 88.72 μmol/L)。其他细胞因子分析均在正常范围内。细胞应激(p38的激活)存在于糖鞘苷(GluSph)积累较高的儿童中。此外,所有受试者LC3-II均显著降低(平均88%,标准差9%,范围77- 98%),表明自噬通量降低。讨论:我们已经在遗传上证实有高谢病高风险的症状前幼儿中发现了炎症与自噬通量抑制的存在。这些发现有助于了解戈谢病的早期病程,并支持通过新生儿筛查确定的高危个体的管理。包括特异性酶替代或其他手段来解决炎症或自噬的治疗干预可以延迟或预防症状性疾病的发生和随之而来的残疾。需要进一步的临床研究来探索这些可能性。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Molecular Genetics and Metabolism Reports
Biochemistry, Genetics and Molecular Biology-Endocrinology
CiteScore
4.00
自引率
5.30%
发文量
105
审稿时长
33 days
期刊介绍:
Molecular Genetics and Metabolism Reports is an open access journal that publishes molecular and metabolic reports describing investigations that use the tools of biochemistry and molecular biology for studies of normal and diseased states. In addition to original research articles, sequence reports, brief communication reports and letters to the editor are considered.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


