Andreina Urquiola Hernández, Christophe Guyeux, Adrien Nicolaï* and Patrick Senet,
{"title":"Molecular Dynamics of Peptide Sequencing through MoS2 Solid-State Nanopores for Binary Encoding Applications","authors":"Andreina Urquiola Hernández, Christophe Guyeux, Adrien Nicolaï* and Patrick Senet, ","doi":"10.1021/acs.jpcb.4c0627710.1021/acs.jpcb.4c06277","DOIUrl":null,"url":null,"abstract":"<p >Biological peptides have emerged as promising candidates for data storage applications due to their versatility and programmability. Recent advances in peptide synthesis and sequencing technologies have enabled the development of peptide-based data storage systems for realizing novel information storage technologies with enhanced capacity, durability, and data access speeds. In this study, we performed coarse-grained peptide sequencing of 12 distinct sequences through single-layer MoS<sub>2</sub> solid-state nanopores (SSNs) using molecular dynamics (MD). Peptide sequences were composed of 1 positively charged, 1 negatively charged, and 4 neutral amino acids, with the position of amino acids in the sequence being shuffled to generate all possible configurations. From MD, the goal was to evaluate the efficiency of these peptide sequences to encode binary information based on ionic current traces monitored during their passage through the SSNs. Classification approaches using LightGBM were trained and tested to analyze different sequence factors such as the position of amino acids or the spacing between charged amino acids in the sequences. Our findings reveal the presence of two distinct groups of sequences determined by the relative position of the positively charged amino acid compared to the negatively charged amino acid. Furthermore, we observe a strong correlation between discrimination accuracy and the separation in the sequence between charged amino acids, depending on the number of adjacent neutral amino acids between them. Finally, MD allowed us to establish the nonlinear relationship between amino acid positions inside the pore (called sequence motifs) and fluctuations in ionic current traces to discriminate false positives and to enable effective training of machine learning classification algorithms. These very promising results emphasized the best approaches to design peptide sequences as building blocks for molecular data storage. Finally, this study highlights the potential of the proposed approach for designing peptide sequence combinations that could help the development of efficient, scalable, and reliable molecular data storage solutions, with future research focused on encoding longer binary chains to enhance storage capacity and support the goal of stable, energy-free biological systems.</p>","PeriodicalId":60,"journal":{"name":"The Journal of Physical Chemistry B","volume":"129 1","pages":"96–110 96–110"},"PeriodicalIF":2.9000,"publicationDate":"2024-12-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry B","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpcb.4c06277","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
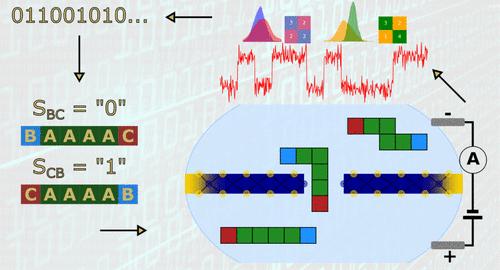
Biological peptides have emerged as promising candidates for data storage applications due to their versatility and programmability. Recent advances in peptide synthesis and sequencing technologies have enabled the development of peptide-based data storage systems for realizing novel information storage technologies with enhanced capacity, durability, and data access speeds. In this study, we performed coarse-grained peptide sequencing of 12 distinct sequences through single-layer MoS2 solid-state nanopores (SSNs) using molecular dynamics (MD). Peptide sequences were composed of 1 positively charged, 1 negatively charged, and 4 neutral amino acids, with the position of amino acids in the sequence being shuffled to generate all possible configurations. From MD, the goal was to evaluate the efficiency of these peptide sequences to encode binary information based on ionic current traces monitored during their passage through the SSNs. Classification approaches using LightGBM were trained and tested to analyze different sequence factors such as the position of amino acids or the spacing between charged amino acids in the sequences. Our findings reveal the presence of two distinct groups of sequences determined by the relative position of the positively charged amino acid compared to the negatively charged amino acid. Furthermore, we observe a strong correlation between discrimination accuracy and the separation in the sequence between charged amino acids, depending on the number of adjacent neutral amino acids between them. Finally, MD allowed us to establish the nonlinear relationship between amino acid positions inside the pore (called sequence motifs) and fluctuations in ionic current traces to discriminate false positives and to enable effective training of machine learning classification algorithms. These very promising results emphasized the best approaches to design peptide sequences as building blocks for molecular data storage. Finally, this study highlights the potential of the proposed approach for designing peptide sequence combinations that could help the development of efficient, scalable, and reliable molecular data storage solutions, with future research focused on encoding longer binary chains to enhance storage capacity and support the goal of stable, energy-free biological systems.
期刊介绍:
An essential criterion for acceptance of research articles in the journal is that they provide new physical insight. Please refer to the New Physical Insights virtual issue on what constitutes new physical insight. Manuscripts that are essentially reporting data or applications of data are, in general, not suitable for publication in JPC B.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


