Incorporation of Ligand Charge and Metal Oxidation State Considerations into the Computational Solvent Removal and Activation of Experimental Crystal Structures Preceding Molecular Simulation
Marco Gibaldi, Anna Kapeliukha, Andrew White and Tom K. Woo*,
{"title":"Incorporation of Ligand Charge and Metal Oxidation State Considerations into the Computational Solvent Removal and Activation of Experimental Crystal Structures Preceding Molecular Simulation","authors":"Marco Gibaldi, Anna Kapeliukha, Andrew White and Tom K. Woo*, ","doi":"10.1021/acs.jcim.4c0189710.1021/acs.jcim.4c01897","DOIUrl":null,"url":null,"abstract":"<p >Efficient computational screenings are integral to materials discovery in highly sought-after gas adsorption and storage applications, such as CO<sub>2</sub> capture. Preprocessing techniques have been developed to render experimental crystal structures suitable for molecular simulations by mimicking experimental activation protocols, particularly residual solvent removal. Current accounts examining these preprocessed materials databases indicate the presence of assorted structural errors introduced by solvent removal and preprocessing, including improper elimination of charge-balancing ions and ligands. Here, we address the need for a reliable experimental crystal structure preprocessing protocol by introducing a novel solvent removal method, which we call SAMOSA, that is informed by systematic ligand charge and metal oxidation state calculations. A robust set of solvent removal criteria is outlined, which identifies solvent molecules and counterions without predefined molecule lists or significant reliance on experimental chemical information. Validation results against popular metal–organic framework (MOF) databases suggest that this method observes significant performance improvements regarding the retention of charged ligands and recognition of charged frameworks. SAMOSA enhances structure fidelity with respect to the original material as-synthesized, thereby representing a powerful tool in computational materials database curation and preprocessing for molecular simulation. The source code is accessible at https://github.com/uowoolab/SAMOSA.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"65 1","pages":"275–287 275–287"},"PeriodicalIF":5.3000,"publicationDate":"2024-12-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.4c01897","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
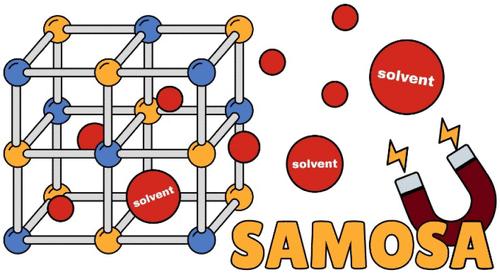
Efficient computational screenings are integral to materials discovery in highly sought-after gas adsorption and storage applications, such as CO2 capture. Preprocessing techniques have been developed to render experimental crystal structures suitable for molecular simulations by mimicking experimental activation protocols, particularly residual solvent removal. Current accounts examining these preprocessed materials databases indicate the presence of assorted structural errors introduced by solvent removal and preprocessing, including improper elimination of charge-balancing ions and ligands. Here, we address the need for a reliable experimental crystal structure preprocessing protocol by introducing a novel solvent removal method, which we call SAMOSA, that is informed by systematic ligand charge and metal oxidation state calculations. A robust set of solvent removal criteria is outlined, which identifies solvent molecules and counterions without predefined molecule lists or significant reliance on experimental chemical information. Validation results against popular metal–organic framework (MOF) databases suggest that this method observes significant performance improvements regarding the retention of charged ligands and recognition of charged frameworks. SAMOSA enhances structure fidelity with respect to the original material as-synthesized, thereby representing a powerful tool in computational materials database curation and preprocessing for molecular simulation. The source code is accessible at https://github.com/uowoolab/SAMOSA.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


