Michel V. Heinz, Emma Gorgas, Nicole Maser, Arne Lüchow
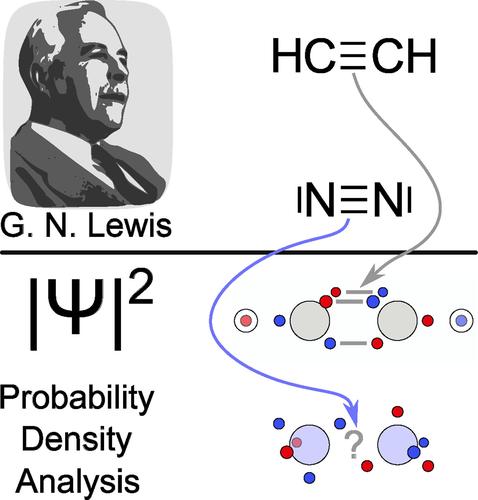
{"title":"Probability Density Analysis Reveals Substantial Differences Between the Dinitrogen and Acetylene Triple Bonds","authors":"Michel V. Heinz, Emma Gorgas, Nicole Maser, Arne Lüchow","doi":"10.1002/jcc.70037","DOIUrl":null,"url":null,"abstract":"<p>In earlier publications, it was shown that the electron positions maximizing the probability density <span></span><math>\n <semantics>\n <mrow>\n <msup>\n <mfenced>\n <mi>Ψ</mi>\n </mfenced>\n <mn>2</mn>\n </msup>\n </mrow>\n <annotation>$$ {\\left|\\Psi \\right|}^2 $$</annotation>\n </semantics></math> resemble the Lewis structures for most small molecules. While this holds for the triple bond in acetylene, this is not the case for the triple bond in dinitrogen. Because of recent advances in studying the topology of wave functions, this peculiar case is revisited. In this work, the dinitrogen wave function is analyzed and compared to that of acetylene. Significant differences of the electron positions maximizing <span></span><math>\n <semantics>\n <mrow>\n <msup>\n <mfenced>\n <mi>Ψ</mi>\n </mfenced>\n <mn>2</mn>\n </msup>\n </mrow>\n <annotation>$$ {\\left|\\Psi \\right|}^2 $$</annotation>\n </semantics></math> are uncovered and explained by the presence of hydrogen atoms in acetylene and by electron arrangements resulting from calculations of the nitrogen and carbon atoms. Moreover, insights into the electron delocalization of both molecules are gained by investigating electron exchange paths. Considering the different chemical behaviors of dinitrogen and acetylene, these differences should be expected.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 4","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2025-02-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.70037","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70037","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
In earlier publications, it was shown that the electron positions maximizing the probability density resemble the Lewis structures for most small molecules. While this holds for the triple bond in acetylene, this is not the case for the triple bond in dinitrogen. Because of recent advances in studying the topology of wave functions, this peculiar case is revisited. In this work, the dinitrogen wave function is analyzed and compared to that of acetylene. Significant differences of the electron positions maximizing are uncovered and explained by the presence of hydrogen atoms in acetylene and by electron arrangements resulting from calculations of the nitrogen and carbon atoms. Moreover, insights into the electron delocalization of both molecules are gained by investigating electron exchange paths. Considering the different chemical behaviors of dinitrogen and acetylene, these differences should be expected.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


