{"title":"Rational Design for Antioxidant Diphenylamine Derivatives Using Quantitative Structure–Activity Relationships and Quantum Mechanics Calculations","authors":"Ayokanmi Joseph Aremu, Phiphob Naweephattana, Ismail Dwi Putra, Borwornlak Toopradab, Phornphimon Maitarad, Thanyada Rungrotmongkol","doi":"10.1002/jcc.70055","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>Diphenylamine (DPA) derivatives, used as antioxidants in rubber-based products, inhibit autoxidation by donating hydrogen atoms to peroxyl radicals. Octanol–water partition coefficient (LogK<sub>ow</sub>), an antioxidant index, helps predict their distribution in hydrophobic polymer matrices. Therefore, this study aimed to investigate the relationship between the structure of DPA derivatives and their antioxidant activities, using machine learning with quantitative structure–activity relationships (QSAR) and quantum mechanics (QM). The structure of DPA derivatives was optimized using Density Functional Theory and analyzed for molecular properties. The QSAR models were trained using important descriptors identified through permutation importance. Among the models developed, the Gradient Boosting Regressor (GBR) showed the best performance, with <i>R</i><sup>2</sup> of 0.983 and root mean square error (RMSE) of 0.642 for the test set. SHAP analysis revealed that molecular weight and electronic properties significantly influenced LogK<sub>ow</sub> predictions. For instance, a higher molecular weight was associated with increased LogK<sub>ow</sub>, and a higher positive charge of C2 correlated with higher LogK<sub>ow</sub> predictions. Consequently, the two potent compounds (D1 and D2) were designed based on QSAR model guidance. The GBR model predicted LogK<sub>ow</sub> values of 9.789 and 7.102 for D1 and D2, respectively, which are higher than the training compounds in the model. To gain molecular insight, the quantum chemical calculations with M062X/6–311++G(d,p)//M062X/6-31G(d,p) were performed to investigate the bond dissociation enthalpy (BDE). The results showed that D1 (79.50 kcal/mol) and D2 (72.43 kcal/mol) exhibited lower BDEs than the reference compounds, suggesting that the designed compounds have the potential for enhanced antioxidant activity. In addition, the antioxidant reaction mechanism was studied by using M062X/6–311++G(d,p)//M062X/6-31G(d,p) which found that the hydrogen atom transfer is the key step, where D1 and D2 showed activation energy barriers of 10.38 and 6.29 kcal/mol, respectively, compared to reference compounds of R3 (10.39 kcal/mol), R1 (10.40 kcal/mol), and R2 (18.26 kcal/mol). Therefore, our findings demonstrate that integrating QSAR with quantum chemical calculations can effectively guide the design of DPA derivatives with improved antioxidant properties.</p>\n </div>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 4","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2025-02-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70055","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
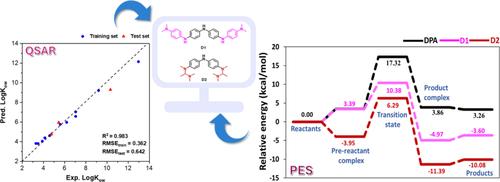
Diphenylamine (DPA) derivatives, used as antioxidants in rubber-based products, inhibit autoxidation by donating hydrogen atoms to peroxyl radicals. Octanol–water partition coefficient (LogKow), an antioxidant index, helps predict their distribution in hydrophobic polymer matrices. Therefore, this study aimed to investigate the relationship between the structure of DPA derivatives and their antioxidant activities, using machine learning with quantitative structure–activity relationships (QSAR) and quantum mechanics (QM). The structure of DPA derivatives was optimized using Density Functional Theory and analyzed for molecular properties. The QSAR models were trained using important descriptors identified through permutation importance. Among the models developed, the Gradient Boosting Regressor (GBR) showed the best performance, with R2 of 0.983 and root mean square error (RMSE) of 0.642 for the test set. SHAP analysis revealed that molecular weight and electronic properties significantly influenced LogKow predictions. For instance, a higher molecular weight was associated with increased LogKow, and a higher positive charge of C2 correlated with higher LogKow predictions. Consequently, the two potent compounds (D1 and D2) were designed based on QSAR model guidance. The GBR model predicted LogKow values of 9.789 and 7.102 for D1 and D2, respectively, which are higher than the training compounds in the model. To gain molecular insight, the quantum chemical calculations with M062X/6–311++G(d,p)//M062X/6-31G(d,p) were performed to investigate the bond dissociation enthalpy (BDE). The results showed that D1 (79.50 kcal/mol) and D2 (72.43 kcal/mol) exhibited lower BDEs than the reference compounds, suggesting that the designed compounds have the potential for enhanced antioxidant activity. In addition, the antioxidant reaction mechanism was studied by using M062X/6–311++G(d,p)//M062X/6-31G(d,p) which found that the hydrogen atom transfer is the key step, where D1 and D2 showed activation energy barriers of 10.38 and 6.29 kcal/mol, respectively, compared to reference compounds of R3 (10.39 kcal/mol), R1 (10.40 kcal/mol), and R2 (18.26 kcal/mol). Therefore, our findings demonstrate that integrating QSAR with quantum chemical calculations can effectively guide the design of DPA derivatives with improved antioxidant properties.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


