Evaluation of the resistome and gut microbiome composition of hospitalized patients in a health unit of southern Brazil coming from a high animal husbandry production region.
Elisa Pires Coltro, Lucas Cafferati Beltrame, Caroline Ribeiro da Cunha, Caetana Paes Zamparette, Clarissa Feltrin, Vilmar Benetti Filho, Patrícia de Almeida Vanny, Sérgio Beduschi Filho, Taíse Costa Ribeiro Klein, Mara Cristina Scheffer, Jussara Kasuko Palmeiro, Glauber Wagner, Thaís Cristine Marques Sincero, Carlos Rodrigo Zárate-Bladés
{"title":"Evaluation of the resistome and gut microbiome composition of hospitalized patients in a health unit of southern Brazil coming from a high animal husbandry production region.","authors":"Elisa Pires Coltro, Lucas Cafferati Beltrame, Caroline Ribeiro da Cunha, Caetana Paes Zamparette, Clarissa Feltrin, Vilmar Benetti Filho, Patrícia de Almeida Vanny, Sérgio Beduschi Filho, Taíse Costa Ribeiro Klein, Mara Cristina Scheffer, Jussara Kasuko Palmeiro, Glauber Wagner, Thaís Cristine Marques Sincero, Carlos Rodrigo Zárate-Bladés","doi":"10.3389/frabi.2024.1489356","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Antimicrobial resistance (AMR) poses a significant threat to global public health. The One Health approach, which integrates human, animal, and environmental health, highlights the roles of agricultural and hospital settings in the propagation of AMR. This study aimed to analyze the resistome and gut microbiome composition of individuals from a high-intensity animal husbandry area in the western region of Santa Catarina, Southern Brazil, who were subsequently admitted to the University Hospital in the city of Florianopolis, located in the eastern part of the same state.</p><p><strong>Methods: </strong>Rectal swab samples were collected upon admission and discharge. Metagenomic sequencing and resistome analysis were employed to identify antimicrobial resistance genes (ARGs) and their associated bacterial taxa. Additionally, the impact of the hospital environment on the resistome and microbiome profiles of these patients was assessed.</p><p><strong>Results: </strong>A total of 247 genetic elements related to AMR were identified, with 66.4% of these elements present in both admission and discharge samples. Aminoglycoside resistance genes were the most prevalent, followed by resistance genes for tetracyclines and lincosamides. Notably, unique resistance genes, including <i>dfrF</i> and mutations in <i>gyrB</i>, were identified at discharge. ARGs were associated with 55 bacterial species, with <i>Lactobacillus fermentum</i>, harboring the ermB gene. (MLSB), detected in both admission and discharge samples. The most prevalent bacterial families included <i>Mycobacteriaceae</i>, Enterobacteriaceae, and <i>Bacteroidaceae</i>. Among these, <i>Mycobacteriaceae</i> was the most abundant, with ARGs primarily associated with mutations in the 16S rRNA gene, RNA polymerase subunits, and gyrases.</p><p><strong>Discussion: </strong>The study revealed a high prevalence of genes related to aminoglycoside and tetracycline resistance, with a notable increase in certain resistance determinants at discharge, likely influenced by extended antimicrobial use. The presence of <i>mcr</i> genes, associated with colistin resistance, in both admission and discharge samples from a single patient highlights a concerning trend in AMR, particularly in relation to animal husbandry. These findings underscore the substantial impact of antimicrobial use on resistance development and the complex dynamics of the resistome in hospital settings. They also emphasize the influence of local factors, such as intensive animal production, on resistance patterns and advocate for ongoing surveillance and policy development to manage multidrug-resistant bacteria eVectively.</p>","PeriodicalId":73065,"journal":{"name":"Frontiers in antibiotics","volume":"3 ","pages":"1489356"},"PeriodicalIF":0.0000,"publicationDate":"2025-01-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11782142/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in antibiotics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/frabi.2024.1489356","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction: Antimicrobial resistance (AMR) poses a significant threat to global public health. The One Health approach, which integrates human, animal, and environmental health, highlights the roles of agricultural and hospital settings in the propagation of AMR. This study aimed to analyze the resistome and gut microbiome composition of individuals from a high-intensity animal husbandry area in the western region of Santa Catarina, Southern Brazil, who were subsequently admitted to the University Hospital in the city of Florianopolis, located in the eastern part of the same state.
Methods: Rectal swab samples were collected upon admission and discharge. Metagenomic sequencing and resistome analysis were employed to identify antimicrobial resistance genes (ARGs) and their associated bacterial taxa. Additionally, the impact of the hospital environment on the resistome and microbiome profiles of these patients was assessed.
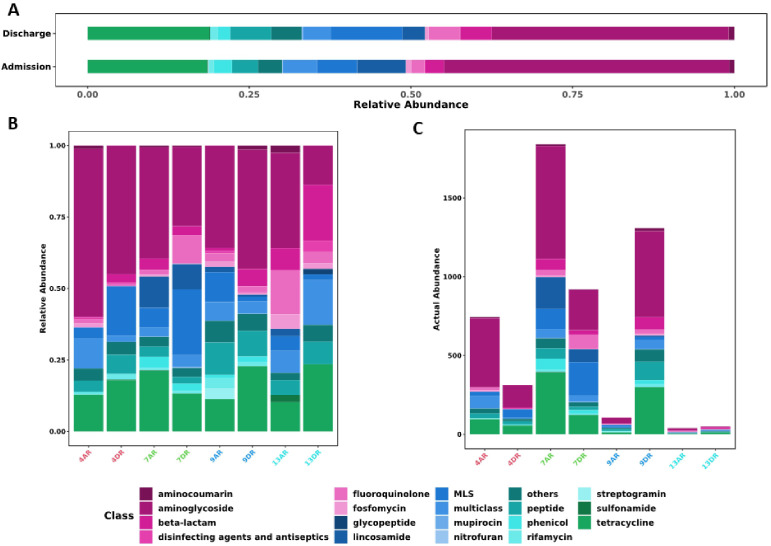
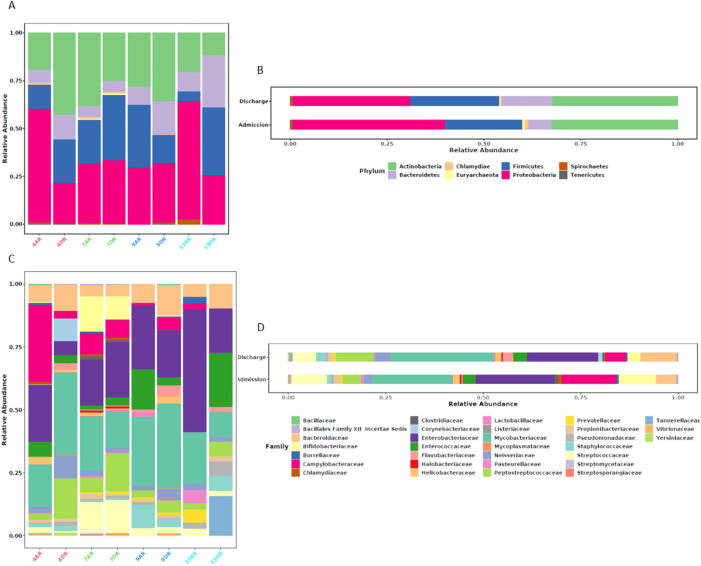
Results: A total of 247 genetic elements related to AMR were identified, with 66.4% of these elements present in both admission and discharge samples. Aminoglycoside resistance genes were the most prevalent, followed by resistance genes for tetracyclines and lincosamides. Notably, unique resistance genes, including dfrF and mutations in gyrB, were identified at discharge. ARGs were associated with 55 bacterial species, with Lactobacillus fermentum, harboring the ermB gene. (MLSB), detected in both admission and discharge samples. The most prevalent bacterial families included Mycobacteriaceae, Enterobacteriaceae, and Bacteroidaceae. Among these, Mycobacteriaceae was the most abundant, with ARGs primarily associated with mutations in the 16S rRNA gene, RNA polymerase subunits, and gyrases.
Discussion: The study revealed a high prevalence of genes related to aminoglycoside and tetracycline resistance, with a notable increase in certain resistance determinants at discharge, likely influenced by extended antimicrobial use. The presence of mcr genes, associated with colistin resistance, in both admission and discharge samples from a single patient highlights a concerning trend in AMR, particularly in relation to animal husbandry. These findings underscore the substantial impact of antimicrobial use on resistance development and the complex dynamics of the resistome in hospital settings. They also emphasize the influence of local factors, such as intensive animal production, on resistance patterns and advocate for ongoing surveillance and policy development to manage multidrug-resistant bacteria eVectively.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


