Electronic and Optical Properties of Highly Complex Ga2O3 and In2O3 Polymorphs Using Approximate Quasiparticle DFT + A – 1/2
IF 3.3
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
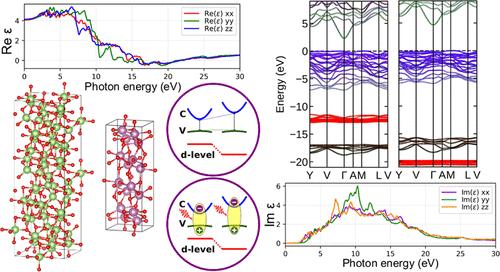
Ga2O3 and In2O3 are among the most important wide-bandgap semiconductors for transparent electronics and ultraviolet optoelectronics. Their pronounced polymorphism necessitates a deeper understanding. In addition to assessing the stability of specific crystal structures, the central goal is to investigate the variation of material properties as a function of the actual crystal structure. The underlying atomic geometries are determined through total energy optimizations within density functional theory (DFT) using the AM05 exchange-correlation functional. To account for excitation effects in electronic systems, the formation of quasiparticles, and the underestimation of band gaps, we employ the fast, efficient, but approximate DFT + A – 1/2 method. This approach accurately predicts fundamental gaps, interband transition energies, and d-level positions for five Ga2O3 and five In2O3 polymorphs, even for structures with up to 160 atoms in the unit cell. The resulting electronic structures are further used to predict dielectric and optical spectra. The effective band masses and dielectric tensors are subsequently employed to estimate the binding energies of band-edge excitons. All results are discussed in the context of polymorph geometry and symmetry, and they are compared with available experimental and theoretical data.
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


