Nanhao Chen, Guodong Rao, Lizhi Tao, R David Britt, Lee-Ping Wang
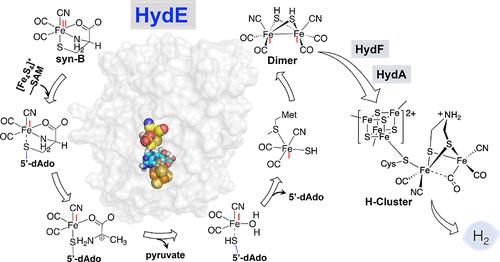
{"title":"HydE Catalytic Mechanism Is Powered by a Radical Relay with Redox-Active Fe(I)-Containing Intermediates.","authors":"Nanhao Chen, Guodong Rao, Lizhi Tao, R David Britt, Lee-Ping Wang","doi":"10.1021/jacs.4c12668","DOIUrl":null,"url":null,"abstract":"<p><p>[FeFe]-hydrogenases are enzymes that catalyze the redox interconversion of H<sup>+</sup> and H<sub>2</sub> using a six-iron active site, known as the H-cluster, which consists of a structurally unique [2Fe]<sub><i>H</i></sub> subcluster linked to a [4Fe-4S]<sub><i>H</i></sub> subcluster. A set of enzymes, HydG, HydE, and HydF, are responsible for the biosynthesis of the [2Fe]<sub><i>H</i></sub> subcluster. Among them, it is well established that HydG cleaves tyrosine into CO and CN<sup>-</sup> and forms a mononuclear [Fe(II)(Cys)(CO)<sub>2</sub>(CN)] complex. Recent work using EPR spectroscopy and X-ray crystallography show that HydE uses this organometallic Fe complex as its native substrate. The low spin Fe(II) center is reduced into an adenosylated Fe(I) species, which is proposed to form an Fe(I)Fe(I) dimer within HydE. The highly unusual transformation catalyzed by HydE draws interest in both biochemistry and organometallic chemistry. Due to the instability of the substrate, the intermediates, and the proposed product, experimental characterization of the detailed HydE mechanism and its final product is challenging. Herein, the catalytic mechanism of HydE is studied using hybrid quantum mechanics/molecular mechanics (QM/MM) molecular dynamics simulations. A radical relay mechanism was found for the cleavage of the cysteine S-Cβ bond that is energetically favored with respect to a closed-shell mechanism involving unconventional proton transfer. In addition, we propose a pathway for the dimerization of two Fe(I) complexes within the HydE hydrophobic cavity, which is consistent with the recent experimental result that HydF can perform [FeFe]-hydrogenase maturation with a synthetic dimer complex as the substrate. These simulation results take us further down the path to a more complete understanding of these enzymes that synthesize one of Nature's most efficient energy conversion catalysts.</p>","PeriodicalId":49,"journal":{"name":"Journal of the American Chemical Society","volume":" ","pages":"4800-4809"},"PeriodicalIF":14.4000,"publicationDate":"2025-02-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11826987/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the American Chemical Society","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/jacs.4c12668","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/30 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
[FeFe]-hydrogenases are enzymes that catalyze the redox interconversion of H+ and H2 using a six-iron active site, known as the H-cluster, which consists of a structurally unique [2Fe]H subcluster linked to a [4Fe-4S]H subcluster. A set of enzymes, HydG, HydE, and HydF, are responsible for the biosynthesis of the [2Fe]H subcluster. Among them, it is well established that HydG cleaves tyrosine into CO and CN- and forms a mononuclear [Fe(II)(Cys)(CO)2(CN)] complex. Recent work using EPR spectroscopy and X-ray crystallography show that HydE uses this organometallic Fe complex as its native substrate. The low spin Fe(II) center is reduced into an adenosylated Fe(I) species, which is proposed to form an Fe(I)Fe(I) dimer within HydE. The highly unusual transformation catalyzed by HydE draws interest in both biochemistry and organometallic chemistry. Due to the instability of the substrate, the intermediates, and the proposed product, experimental characterization of the detailed HydE mechanism and its final product is challenging. Herein, the catalytic mechanism of HydE is studied using hybrid quantum mechanics/molecular mechanics (QM/MM) molecular dynamics simulations. A radical relay mechanism was found for the cleavage of the cysteine S-Cβ bond that is energetically favored with respect to a closed-shell mechanism involving unconventional proton transfer. In addition, we propose a pathway for the dimerization of two Fe(I) complexes within the HydE hydrophobic cavity, which is consistent with the recent experimental result that HydF can perform [FeFe]-hydrogenase maturation with a synthetic dimer complex as the substrate. These simulation results take us further down the path to a more complete understanding of these enzymes that synthesize one of Nature's most efficient energy conversion catalysts.
期刊介绍:
The flagship journal of the American Chemical Society, known as the Journal of the American Chemical Society (JACS), has been a prestigious publication since its establishment in 1879. It holds a preeminent position in the field of chemistry and related interdisciplinary sciences. JACS is committed to disseminating cutting-edge research papers, covering a wide range of topics, and encompasses approximately 19,000 pages of Articles, Communications, and Perspectives annually. With a weekly publication frequency, JACS plays a vital role in advancing the field of chemistry by providing essential research.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


