Christopher D Thouvenel, Christopher M Tipton, Yasuhiro Yamazaki, Ting-Ting Zhang, Stacey Rylaarsdam, Jennifer R Hom, Catherine Snead, Chengsong Zhu, Quan-Zhen Li, Yu Nee Lee, Tomoki Kawai, Neshatul Haque, Michael T Zimmermann, Sivasankaran Munusamy Ponnan, Shaun W Jackson, Rich G James, Ignacio Sanz, Luigi D Notarangelo, Troy R Torgerson, Hans D Ochs, David J Rawlings, Eric J Allenspach
{"title":"Hypomorphic RAG2 Deficiency Promotes Selection of Self-Reactive B Cells.","authors":"Christopher D Thouvenel, Christopher M Tipton, Yasuhiro Yamazaki, Ting-Ting Zhang, Stacey Rylaarsdam, Jennifer R Hom, Catherine Snead, Chengsong Zhu, Quan-Zhen Li, Yu Nee Lee, Tomoki Kawai, Neshatul Haque, Michael T Zimmermann, Sivasankaran Munusamy Ponnan, Shaun W Jackson, Rich G James, Ignacio Sanz, Luigi D Notarangelo, Troy R Torgerson, Hans D Ochs, David J Rawlings, Eric J Allenspach","doi":"10.1007/s10875-024-01849-9","DOIUrl":null,"url":null,"abstract":"<p><p>Reduced function or hypomorphic variants in recombination-activating genes (RAG) 1 or 2 result in a broad clinical phenotype including common variable immunodeficiency (CVID) and even adult-onset disease. Milder RAG variants are less characterized. Here we describe the longitudinal course of a milder combined RAG deficiency in 3 of 7 siblings sharing the same RAG2 mutations over a 50-year study. Whole-genome and repertoire sequencing, bacteriophage immunizations, and deep immunophenotyping were used to compare affected and unaffected family members. The clinical phenotype of three affected siblings with hypomorphic RAG deficiency ranged from combined immunodeficiency and early mortality to a late-onset CID with hyper-IgM phenotype. T cells were remarkably similar across affected siblings, yet CDR3 skewing and regulatory T cell defects were not observed. B cell analysis showed elevated unswitched CD27+ and CD21<sup>low</sup> cells as well as features of an autoreactive antibody repertoire and presence of secreted autoantibodies, yet no clinical autoimmunity was present. Most striking was an expanded polyclonal marginal zone-like B cell population (IgM+IgD+CD27+) utilizing the self-reactive unmutated VH4-34 receptor demonstrating that hypomorphic RAG deficiency can promote expansion of self-reactive B cells. This process, however, was not sufficient to trigger clinical autoimmunity. Utilizing multiple approaches, we functionally measured the specific RAG2 variant effects and assessed how selection and secondary triggers altered the BCR repertoire and immunophenotype overtime. Overall, we demonstrate a broad disease spectrum in siblings with identical hypomorphic RAG deficiency, highlighting that phenotypic divergence can result from expansion of IgM + memory B cells.</p>","PeriodicalId":15531,"journal":{"name":"Journal of Clinical Immunology","volume":"45 1","pages":"66"},"PeriodicalIF":5.7000,"publicationDate":"2025-01-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11735530/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Immunology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10875-024-01849-9","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
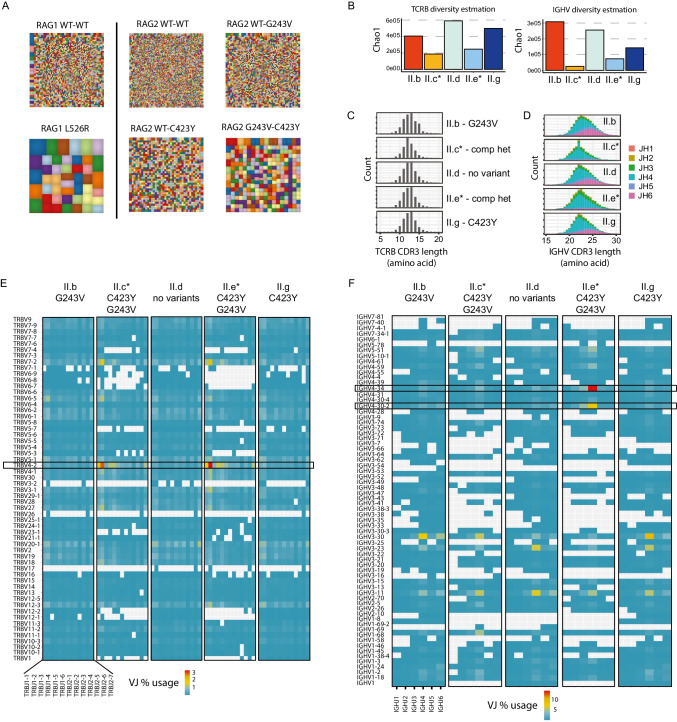
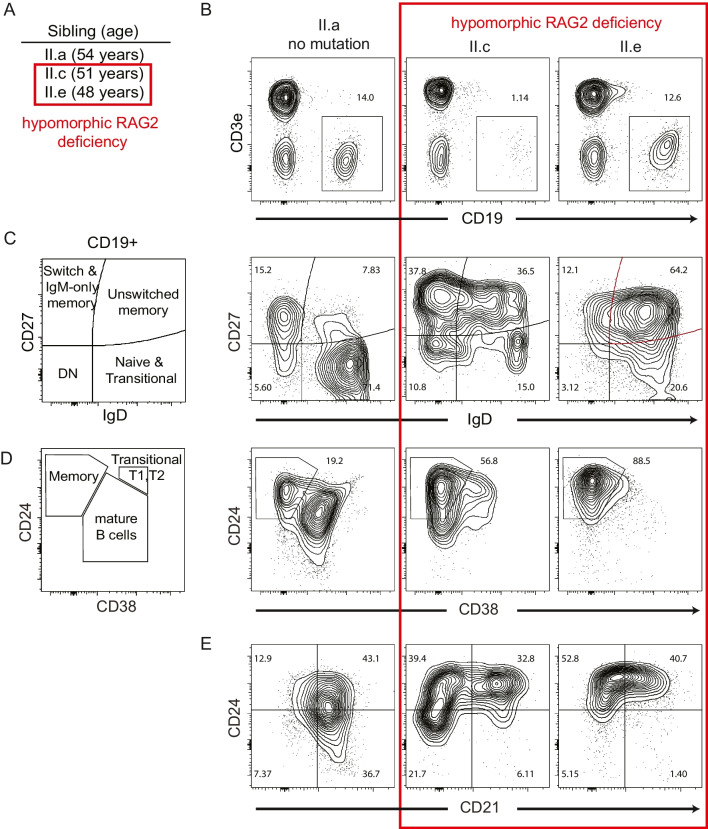
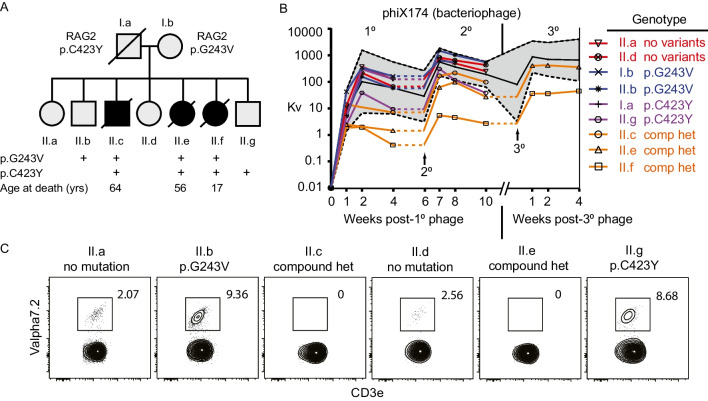
Reduced function or hypomorphic variants in recombination-activating genes (RAG) 1 or 2 result in a broad clinical phenotype including common variable immunodeficiency (CVID) and even adult-onset disease. Milder RAG variants are less characterized. Here we describe the longitudinal course of a milder combined RAG deficiency in 3 of 7 siblings sharing the same RAG2 mutations over a 50-year study. Whole-genome and repertoire sequencing, bacteriophage immunizations, and deep immunophenotyping were used to compare affected and unaffected family members. The clinical phenotype of three affected siblings with hypomorphic RAG deficiency ranged from combined immunodeficiency and early mortality to a late-onset CID with hyper-IgM phenotype. T cells were remarkably similar across affected siblings, yet CDR3 skewing and regulatory T cell defects were not observed. B cell analysis showed elevated unswitched CD27+ and CD21low cells as well as features of an autoreactive antibody repertoire and presence of secreted autoantibodies, yet no clinical autoimmunity was present. Most striking was an expanded polyclonal marginal zone-like B cell population (IgM+IgD+CD27+) utilizing the self-reactive unmutated VH4-34 receptor demonstrating that hypomorphic RAG deficiency can promote expansion of self-reactive B cells. This process, however, was not sufficient to trigger clinical autoimmunity. Utilizing multiple approaches, we functionally measured the specific RAG2 variant effects and assessed how selection and secondary triggers altered the BCR repertoire and immunophenotype overtime. Overall, we demonstrate a broad disease spectrum in siblings with identical hypomorphic RAG deficiency, highlighting that phenotypic divergence can result from expansion of IgM + memory B cells.
期刊介绍:
The Journal of Clinical Immunology publishes impactful papers in the realm of human immunology, delving into the diagnosis, pathogenesis, prognosis, or treatment of human diseases. The journal places particular emphasis on primary immunodeficiencies and related diseases, encompassing inborn errors of immunity in a broad sense, their underlying genotypes, and diverse phenotypes. These phenotypes include infection, malignancy, allergy, auto-inflammation, and autoimmunity. We welcome a broad spectrum of studies in this domain, spanning genetic discovery, clinical description, immunologic assessment, diagnostic approaches, prognosis evaluation, and treatment interventions. Case reports are considered if they are genuinely original and accompanied by a concise review of the relevant medical literature, illustrating how the novel case study advances the field. The instructions to authors provide detailed guidance on the four categories of papers accepted by the journal.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


