André F Rodrigues, Laura Boreggio, Tetiana Lahuta, Fatimunnisa Qadri, Natalia Alenina, Carlos C Barros, Mihail Todiras, Michael Bader
{"title":"Renal damage-induced hepcidin accumulation contributes to anemia in angiotensinogen-deficient mice.","authors":"André F Rodrigues, Laura Boreggio, Tetiana Lahuta, Fatimunnisa Qadri, Natalia Alenina, Carlos C Barros, Mihail Todiras, Michael Bader","doi":"10.1042/CS20241789","DOIUrl":null,"url":null,"abstract":"<p><p>Angiotensin II (Ang II) is the most active peptide hormone produced by the renin-angiotensin system (RAS). Genetic deletion of genes that ultimately restrict Ang II formation has been shown to result in marked anemia in mice. In this study, adult mice with a genetic deletion of the RAS precursor protein angiotensinogen (Agt-KO) were used. Experimental analyses included capillary hematocrit, hemogram, plasma and tissue iron quantifications, expression analyses of genes encoding relevant proteins for body iron homeostasis in different organs, as well as plasma and urine hepcidin quantifications. As previously reported, Agt-KO were anemic with reduced red blood cell counts. Interestingly, we found that they presented microcytic anemia based on the reduced red blood cell volume. In agreement, plasma quantification of iron revealed lower levels of circulating iron in Agt-KO. The major body iron stores, namely in the liver and spleen, were also depleted in the RAS-deficient line. Hepatic hepcidin expression was reduced, as well as one of its major regulators, BMP6, as a result of the iron deficiency. However, plasma hepcidin levels were unexpectedly increased in Agt-KO. We confirm the typical morphological alterations and impaired renal function of Agt-KO and conclude that hepcidin accumulates in the circulation due to the reduced glomerular filtration in Agt-KO, and therefore identified the culprit of iron deficiency in Agt-KO. Collectively, the data demonstrated that the severe anemia developed in RAS-deficient mice is exacerbated by iron deficiency which is secondary to the renal damage-induced hepcidin accumulation in the circulation.</p>","PeriodicalId":10475,"journal":{"name":"Clinical science","volume":" ","pages":""},"PeriodicalIF":7.7000,"publicationDate":"2025-02-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12203995/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical science","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1042/CS20241789","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
引用次数: 0
Abstract
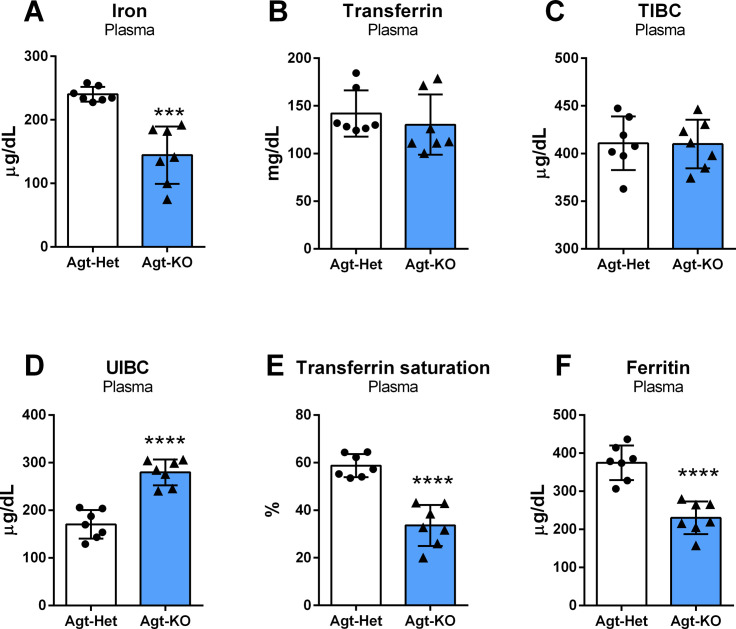
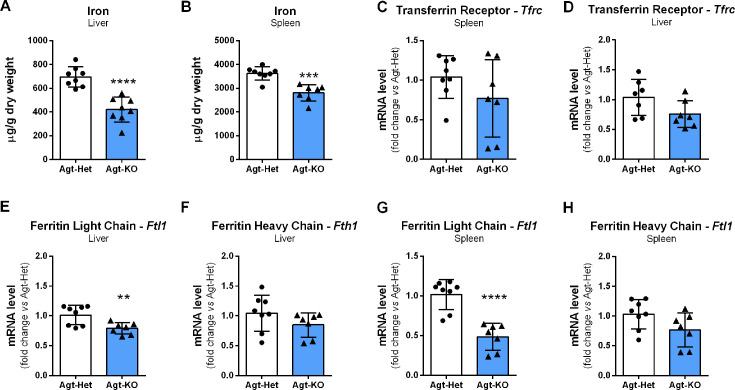
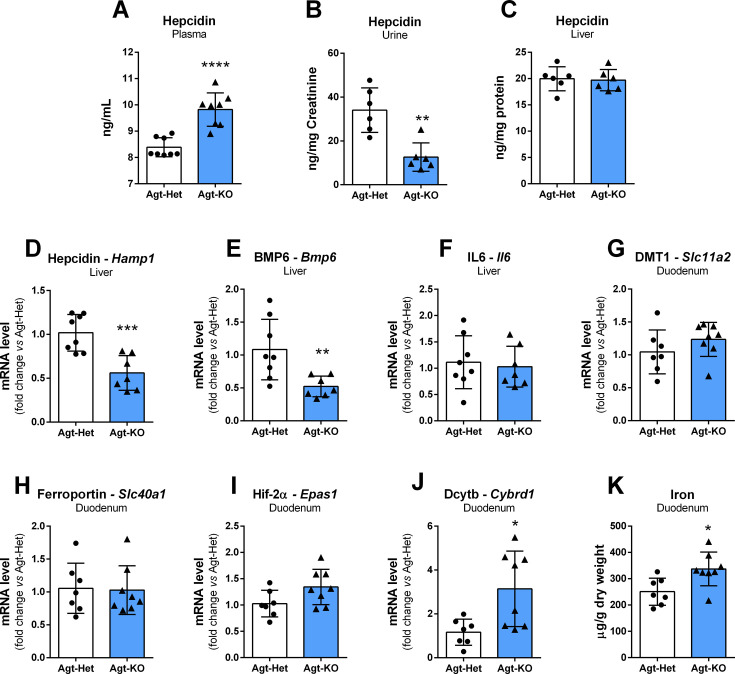
Angiotensin II (Ang II) is the most active peptide hormone produced by the renin-angiotensin system (RAS). Genetic deletion of genes that ultimately restrict Ang II formation has been shown to result in marked anemia in mice. In this study, adult mice with a genetic deletion of the RAS precursor protein angiotensinogen (Agt-KO) were used. Experimental analyses included capillary hematocrit, hemogram, plasma and tissue iron quantifications, expression analyses of genes encoding relevant proteins for body iron homeostasis in different organs, as well as plasma and urine hepcidin quantifications. As previously reported, Agt-KO were anemic with reduced red blood cell counts. Interestingly, we found that they presented microcytic anemia based on the reduced red blood cell volume. In agreement, plasma quantification of iron revealed lower levels of circulating iron in Agt-KO. The major body iron stores, namely in the liver and spleen, were also depleted in the RAS-deficient line. Hepatic hepcidin expression was reduced, as well as one of its major regulators, BMP6, as a result of the iron deficiency. However, plasma hepcidin levels were unexpectedly increased in Agt-KO. We confirm the typical morphological alterations and impaired renal function of Agt-KO and conclude that hepcidin accumulates in the circulation due to the reduced glomerular filtration in Agt-KO, and therefore identified the culprit of iron deficiency in Agt-KO. Collectively, the data demonstrated that the severe anemia developed in RAS-deficient mice is exacerbated by iron deficiency which is secondary to the renal damage-induced hepcidin accumulation in the circulation.
期刊介绍:
Translating molecular bioscience and experimental research into medical insights, Clinical Science offers multi-disciplinary coverage and clinical perspectives to advance human health.
Its international Editorial Board is charged with selecting peer-reviewed original papers of the highest scientific merit covering the broad spectrum of biomedical specialities including, although not exclusively:
Cardiovascular system
Cerebrovascular system
Gastrointestinal tract and liver
Genomic medicine
Infection and immunity
Inflammation
Oncology
Metabolism
Endocrinology and nutrition
Nephrology
Circulation
Respiratory system
Vascular biology
Molecular pathology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


