{"title":"Redox regulation of TRIM28 facilitates neuronal ferroptosis by promoting SUMOylation and inhibiting OPTN-selective autophagic degradation of ACSL4","authors":"Wei Liu, Yufeng Zhu, Wu Ye, Junjun Xiong, Haofan Wang, Yu Gao, Shixue Huang, Yinuo Zhang, Xin Zhou, Xuhui Zhou, Xuhui Ge, Weihua Cai, Xingdong Zheng","doi":"10.1038/s41418-025-01452-4","DOIUrl":null,"url":null,"abstract":"<p>Ferroptosis is one of the cell death programs occurring after spinal cord injury (SCI) and is driven by iron-dependent phospholipid peroxidation. However, little is known about its underlying regulation mechanism. The present study demonstrated that lipid peroxidation was promoted in patients with SCI. Neurons affected by ferroptosis following SCI had a high expression of ferroptotic protein ACSL4. The E3 SUMOylase TRIM28 promoted neuronal ferroptosis by enhancing ACSL4 expression. Genetic deletion of <i>Trim28</i> significantly attenuated neuronal ferroptosis and improved mouse hindlimb motor function following SCI. In contrast, mice with <i>Trim28</i> overexpression demonstrated poor neurological function after SCI, which was attenuated by ferroptosis inhibitor Liproxstatin-1. Mechanistically, TRIM28 bound to ACSL4, promoted SUMO3 modification at lysine (K) 532, and inhibited K63-linked ACSL4 ubiquitination, thereby suppressing OPTN-dependent autophagic degradation. Additionally, SENP3 was identified as the deSUMOylation enzyme that can reverse this process and compete with TRIM28, which was transcriptionally upregulated due to excessive oxidative stress. These data unveiled a mechanism by which TRIM28-mediated SUMOylation regulated neuronal ACSL4 levels and ferroptosis, identified interactions and correlations involved in ACSL4 SUMOylation, ubiquitination, and autophagic degradation, and discovered a positive feedback loop where oxidative stress transcriptionally upregulated <i>Trim28</i>, and conversely TRIM28 promoted ferroptosis and oxidative stress. Notably, screening of the FDA-approved drug library revealed that pharmacological TRIM28/ACSL4 axis interventions with Rutin hydrate inhibited neuronal ferroptosis and improved hindlimb motor function in mice after SCI, thus providing a promising therapeutic strategy for its treatment.</p>","PeriodicalId":9731,"journal":{"name":"Cell Death and Differentiation","volume":"89 1","pages":""},"PeriodicalIF":15.4000,"publicationDate":"2025-01-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell Death and Differentiation","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1038/s41418-025-01452-4","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
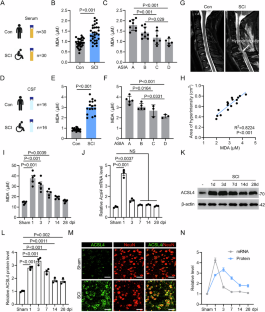
Ferroptosis is one of the cell death programs occurring after spinal cord injury (SCI) and is driven by iron-dependent phospholipid peroxidation. However, little is known about its underlying regulation mechanism. The present study demonstrated that lipid peroxidation was promoted in patients with SCI. Neurons affected by ferroptosis following SCI had a high expression of ferroptotic protein ACSL4. The E3 SUMOylase TRIM28 promoted neuronal ferroptosis by enhancing ACSL4 expression. Genetic deletion of Trim28 significantly attenuated neuronal ferroptosis and improved mouse hindlimb motor function following SCI. In contrast, mice with Trim28 overexpression demonstrated poor neurological function after SCI, which was attenuated by ferroptosis inhibitor Liproxstatin-1. Mechanistically, TRIM28 bound to ACSL4, promoted SUMO3 modification at lysine (K) 532, and inhibited K63-linked ACSL4 ubiquitination, thereby suppressing OPTN-dependent autophagic degradation. Additionally, SENP3 was identified as the deSUMOylation enzyme that can reverse this process and compete with TRIM28, which was transcriptionally upregulated due to excessive oxidative stress. These data unveiled a mechanism by which TRIM28-mediated SUMOylation regulated neuronal ACSL4 levels and ferroptosis, identified interactions and correlations involved in ACSL4 SUMOylation, ubiquitination, and autophagic degradation, and discovered a positive feedback loop where oxidative stress transcriptionally upregulated Trim28, and conversely TRIM28 promoted ferroptosis and oxidative stress. Notably, screening of the FDA-approved drug library revealed that pharmacological TRIM28/ACSL4 axis interventions with Rutin hydrate inhibited neuronal ferroptosis and improved hindlimb motor function in mice after SCI, thus providing a promising therapeutic strategy for its treatment.
期刊介绍:
Mission, vision and values of Cell Death & Differentiation:
To devote itself to scientific excellence in the field of cell biology, molecular biology, and biochemistry of cell death and disease.
To provide a unified forum for scientists and clinical researchers
It is committed to the rapid publication of high quality original papers relating to these subjects, together with topical, usually solicited, reviews, meeting reports, editorial correspondence and occasional commentaries on controversial and scientifically informative issues.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


