Crystal Structure Prediction of Cs–Te with Supervised Machine Learning
IF 2.9
4区 工程技术
Q1 MULTIDISCIPLINARY SCIENCES
引用次数: 0
Abstract
Crystal structure prediction methods aim to determine the ground-state crystal structure for a given material. The vast combinatorial space associated with this problem makes conventional methods computationally prohibitive for routine use. To overcome these limitations, a novel approach combining high-throughput density functional theory calculations with machine learning is proposed. It predicts stable crystal structures within binary and ternary systems by systematically evaluating various structural descriptors and machine learning algorithms. The superiority of models based on atomic coordination environments is shown, with transfer-learned graph neural networks emerging as a particularly promising technique. By validating the proposed method on Cs–Te crystals, its ability to generate stable crystal structures is proved, suggesting its potential for advancing established computational schemes.
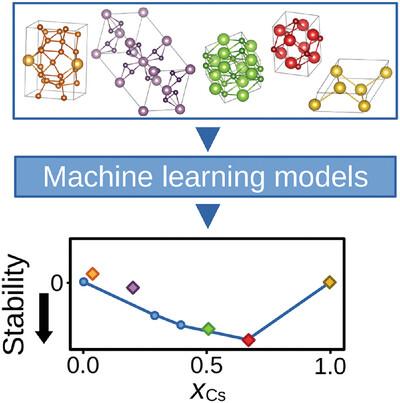
基于监督机器学习的Cs-Te晶体结构预测
晶体结构预测方法的目的是确定给定材料的基态晶体结构。与这个问题相关的巨大组合空间使得传统的计算方法在日常使用中望而却步。为了克服这些限制,提出了一种将高通量密度泛函理论计算与机器学习相结合的新方法。它通过系统地评估各种结构描述符和机器学习算法来预测二元和三元体系中的稳定晶体结构。基于原子配位环境的模型的优越性得到了体现,其中迁移学习图神经网络是一种特别有前途的技术。通过在Cs-Te晶体上验证所提出的方法,证明了其产生稳定晶体结构的能力,表明其具有推进已建立的计算方案的潜力。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Advanced Theory and Simulations
Multidisciplinary-Multidisciplinary
CiteScore
5.50
自引率
3.00%
发文量
221
期刊介绍:
Advanced Theory and Simulations is an interdisciplinary, international, English-language journal that publishes high-quality scientific results focusing on the development and application of theoretical methods, modeling and simulation approaches in all natural science and medicine areas, including:
materials, chemistry, condensed matter physics
engineering, energy
life science, biology, medicine
atmospheric/environmental science, climate science
planetary science, astronomy, cosmology
method development, numerical methods, statistics
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


