Joshua Pandian, Khanh Vu, Jules Tshishimbi Muya, Anna Parker, Christine Mae F. Ancajas, Diomedes Saldana-Greco, Tabitha Yewer, Carol Parish
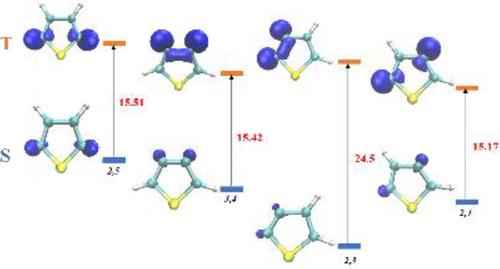
{"title":"A Highly Correlated, Multireference Study of the Lowest Lying Singlet and Triplet States of the Four Thiophene Diradicals","authors":"Joshua Pandian, Khanh Vu, Jules Tshishimbi Muya, Anna Parker, Christine Mae F. Ancajas, Diomedes Saldana-Greco, Tabitha Yewer, Carol Parish","doi":"10.1002/jcc.70044","DOIUrl":null,"url":null,"abstract":"<p>The energies and geometries of the lowest lying singlet and triplet states of the four diradicals formed by removing two H atoms from thiophene have been characterized. We utilized the highly correlated, multireference methods configuration interaction with single and double excitations with and without the Pople correction for size-extensivity (MR-CISD+Q and MR-CISD) and averaged quadratic coupled cluster theory (MR-AQCC). CAS (8,7) and CAS (10,8) active spaces involving σ, σ*, π, and π* orbitals were employed along with the cc-pVDZ and cc-pVTZ basis sets. The larger active space included the two electrons in the nonbonding sp<sup>2</sup> hybrid orbital on sulfur. We find that all didehydro isomers exist as planar, stable ground state singlets. The singlet-triplet (S-T) adiabatic gaps range from 15 to 25 kcal/mol while the vertical splittings are 21–35 kcal/mol. The 2,3 isomer has the lowest absolute ground state singlet energy and the largest adiabatic and vertical S-T splitting. The ground states of the 2,3-, and 2,5-didehydrothiophene isomers are predicted to exhibit the smallest and largest diradical character, respectively, based on their electronic structures, spin densities and bonding analysis. To our knowledge, no experimental excitation energies of any of the didehydrothiophene isomers are available, and our computed MR-AQCC/cc-pVTZ data are believed to be among the most accurate computed results. This extensive study shows a competitive performance between MR-AQCC and MR-CISD+Q.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 3","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2025-01-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.70044","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70044","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
The energies and geometries of the lowest lying singlet and triplet states of the four diradicals formed by removing two H atoms from thiophene have been characterized. We utilized the highly correlated, multireference methods configuration interaction with single and double excitations with and without the Pople correction for size-extensivity (MR-CISD+Q and MR-CISD) and averaged quadratic coupled cluster theory (MR-AQCC). CAS (8,7) and CAS (10,8) active spaces involving σ, σ*, π, and π* orbitals were employed along with the cc-pVDZ and cc-pVTZ basis sets. The larger active space included the two electrons in the nonbonding sp2 hybrid orbital on sulfur. We find that all didehydro isomers exist as planar, stable ground state singlets. The singlet-triplet (S-T) adiabatic gaps range from 15 to 25 kcal/mol while the vertical splittings are 21–35 kcal/mol. The 2,3 isomer has the lowest absolute ground state singlet energy and the largest adiabatic and vertical S-T splitting. The ground states of the 2,3-, and 2,5-didehydrothiophene isomers are predicted to exhibit the smallest and largest diradical character, respectively, based on their electronic structures, spin densities and bonding analysis. To our knowledge, no experimental excitation energies of any of the didehydrothiophene isomers are available, and our computed MR-AQCC/cc-pVTZ data are believed to be among the most accurate computed results. This extensive study shows a competitive performance between MR-AQCC and MR-CISD+Q.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


