Mauricio Guerrero-Montero, Michał Bosy, Christopher D. Cooper
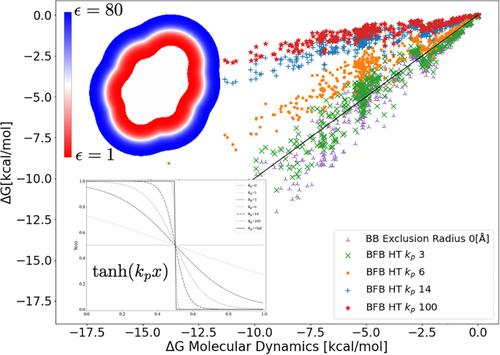
{"title":"Some Challenges of Diffused Interfaces in Implicit-Solvent Models","authors":"Mauricio Guerrero-Montero, Michał Bosy, Christopher D. Cooper","doi":"10.1002/jcc.70036","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>The standard Poisson-Boltzmann (PB) model for molecular electrostatics assumes a sharp variation of the permittivity and salt concentration along the solute-solvent interface. The discontinuous field parameters are not only difficult numerically, but also are not a realistic physical picture, as it forces the dielectric constant and ionic strength of bulk in the near-solute region. An alternative to alleviate some of these issues is to represent the molecular surface as a diffuse interface, however, this also presents challenges. In this work we analyzed the impact of the shape of the interfacial variation of the field parameters in solvation and binding energy. However we used a hyperbolic tangent function <span></span><math>\n <semantics>\n <mrow>\n <mfenced>\n <mrow>\n <mi>tanh</mi>\n <mfenced>\n <mrow>\n <msub>\n <mi>k</mi>\n <mi>p</mi>\n </msub>\n <mi>x</mi>\n </mrow>\n </mfenced>\n </mrow>\n </mfenced>\n </mrow>\n <annotation>$$ \\left(\\tanh \\left({k}_px\\right)\\right) $$</annotation>\n </semantics></math> to couple the internal and external regions, our analysis is valid for other definitions. Our methodology, restricted to the linear PB, was based on a coupled finite element (FEM) and boundary element (BEM) scheme that allowed us to have a special treatment of the permittivity and ionic strength in a bounded FEM region near the interface, while maintaining BEM elsewhere. Our results suggest that the shape of the function (represented by <span></span><math>\n <semantics>\n <mrow>\n <msub>\n <mi>k</mi>\n <mi>p</mi>\n </msub>\n </mrow>\n <annotation>$$ {k}_p $$</annotation>\n </semantics></math>) has a large impact on solvation and binding energy. We saw that high values of <span></span><math>\n <semantics>\n <mrow>\n <msub>\n <mi>k</mi>\n <mi>p</mi>\n </msub>\n </mrow>\n <annotation>$$ {k}_p $$</annotation>\n </semantics></math> induce a high gradient on the interface, to the limit of recovering the sharp jump when <span></span><math>\n <semantics>\n <mrow>\n <msub>\n <mi>k</mi>\n <mi>p</mi>\n </msub>\n <mo>→</mo>\n <mo>∞</mo>\n </mrow>\n <annotation>$$ {k}_p\\to \\infty $$</annotation>\n </semantics></math>, presenting a numerical challenge where careful meshing is key. Using the FreeSolv database to compare with molecular dynamics, our calculations indicate that an optimal value of <span></span><math>\n <semantics>\n <mrow>\n <msub>\n <mi>k</mi>\n <mi>p</mi>\n </msub>\n </mrow>\n <annotation>$$ {k}_p $$</annotation>\n </semantics></math> for solvation energies was around 3. However, more challenging binding free energy tests make this conclusion more difficult, as binding showed to be very sensitive to small variations of <span></span><math>\n <semantics>\n <mrow>\n <msub>\n <mi>k</mi>\n <mi>p</mi>\n </msub>\n </mrow>\n <annotation>$$ {k}_p $$</annotation>\n </semantics></math>. In that case, optimal values of <span></span><math>\n <semantics>\n <mrow>\n <msub>\n <mi>k</mi>\n <mi>p</mi>\n </msub>\n </mrow>\n <annotation>$$ {k}_p $$</annotation>\n </semantics></math> ranged from 2 to 20.</p>\n </div>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 3","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2025-01-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70036","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
The standard Poisson-Boltzmann (PB) model for molecular electrostatics assumes a sharp variation of the permittivity and salt concentration along the solute-solvent interface. The discontinuous field parameters are not only difficult numerically, but also are not a realistic physical picture, as it forces the dielectric constant and ionic strength of bulk in the near-solute region. An alternative to alleviate some of these issues is to represent the molecular surface as a diffuse interface, however, this also presents challenges. In this work we analyzed the impact of the shape of the interfacial variation of the field parameters in solvation and binding energy. However we used a hyperbolic tangent function to couple the internal and external regions, our analysis is valid for other definitions. Our methodology, restricted to the linear PB, was based on a coupled finite element (FEM) and boundary element (BEM) scheme that allowed us to have a special treatment of the permittivity and ionic strength in a bounded FEM region near the interface, while maintaining BEM elsewhere. Our results suggest that the shape of the function (represented by ) has a large impact on solvation and binding energy. We saw that high values of induce a high gradient on the interface, to the limit of recovering the sharp jump when , presenting a numerical challenge where careful meshing is key. Using the FreeSolv database to compare with molecular dynamics, our calculations indicate that an optimal value of for solvation energies was around 3. However, more challenging binding free energy tests make this conclusion more difficult, as binding showed to be very sensitive to small variations of . In that case, optimal values of ranged from 2 to 20.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


