Simplified Multireference Coupled-Cluster Methods: Hybrid Approaches With Averaged Coupled Pair Theories
IF 3.4
3区 化学
Q2 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
Abstract
We define an approximation to the internally contracted multireference coupled-cluster method with single and double excitations by a hybrid approach. The rationale is to treat the external pair energy contributions by the coupled-cluster method, which provides accurate results for a large part of the correlation energy while being tractable as the involved pair cluster operators commute. For the internal and semi-internal contributions, for which the coupled-cluster part becomes involved due to non-commuting operators, a linearized approach based on the coupled-electron pair approximation (CEPA) is used. For the latter, the CEPA(0) method, the averaged coupled pair functional (ACPF), the averaged quadratic coupled-cluster (AQCC) method, and the averaged CEPA method are tested. We test the methods concerning size consistency, potential energy curves for C2, N2, CN, and O3 and for the singlet-triplet splitting of ortho-, meta-, and para-benzynes. Our results show that AQCC provides the most accurate results and stable performance. The main drawback of the method is that it shows small violations of size consistency.
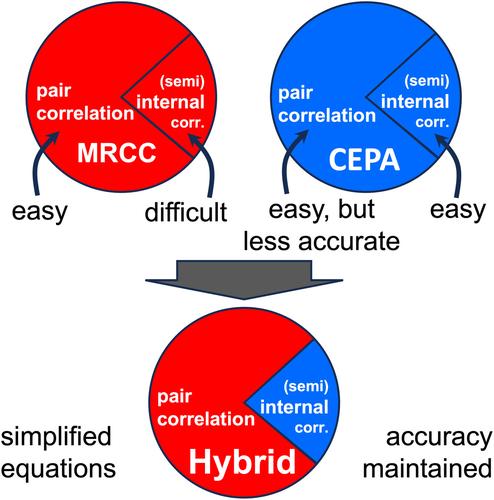
简化多参考耦合聚类方法:平均耦合对理论的混合方法
用一种混合方法定义了具有单激励和双激励的内收缩多参考耦合簇方法的近似。其基本原理是通过耦合聚类方法处理外部对能量的贡献,该方法为大部分相关能量提供了准确的结果,同时在所涉及的对聚类算子交换时易于处理。对于由于非交换算子而涉及到耦合簇部分的内部和半内部贡献,采用了基于耦合电子对近似(CEPA)的线性化方法。对于后者,测试了CEPA(0)方法、平均耦合对泛函(ACPF)方法、平均二次耦合簇(AQCC)方法和平均CEPA方法。我们测试了C2、N2、CN和O3的尺寸一致性、势能曲线以及邻苯、间苯和对苯的单重态-三重态分裂。实验结果表明,AQCC的结果最准确,性能稳定。该方法的主要缺点是它显示了大小一致性的小违规。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

CiteScore
6.60
自引率
3.30%
发文量
247
审稿时长
1.7 months
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


