Anisley Valenciaga, Pamela Brock, Benjamin O'Donnell, Steven W Ing
{"title":"Diagnosing Hypoparathyroidism, Sensorineural Deafness, and Renal Dysplasia Syndrome and a Novel <i>GATA3</i> Variant.","authors":"Anisley Valenciaga, Pamela Brock, Benjamin O'Donnell, Steven W Ing","doi":"10.1210/jcemcr/luae246","DOIUrl":null,"url":null,"abstract":"<p><p>Hypoparathyroidism (hypoPTH), sensorineural deafness, and renal dysplasia (HDR) syndrome is a rare autosomal dominant condition with approximately 200 cases published. HDR syndrome is caused by variants of GATA binding protein 3 gene (<i>GATA3</i>), which encodes a transcription factor, with multiple types of <i>GATA3</i> variants reported. We present the case of a 76-year-old woman who was diagnosed with hypoPTH when she was aged 40 years and transferred care to our institution. Further history elucidated presence of deafness at age 1 year and chronic kidney disease with a left atrophic kidney diagnosed in her 60 seconds. Genetic testing identified a novel <i>GATA3</i> missense variant of unknown significance (c.791G > A, p.Cys264Tyr). There was no family history of hypoPTH, deafness, or renal disease, which might indicate incomplete penetrance or de novo mutation. Advanced modeling of protein sequence and biophysical properties predicts abnormal protein function, suggesting possible pathogenicity. In addition, a likely pathogenic variant in the same amino acid was previously described in a patient with HDR, supporting the in silico prediction of pathogenicity in our patient's variant. Syndromic hypoPTH should be considered in patients even if presenting later in life with presumed chronic isolated conditions. Genetic testing can guide further disease screening and family testing when appropriate.</p>","PeriodicalId":73540,"journal":{"name":"JCEM case reports","volume":"3 1","pages":"luae246"},"PeriodicalIF":0.0000,"publicationDate":"2025-01-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11735463/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JCEM case reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1210/jcemcr/luae246","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
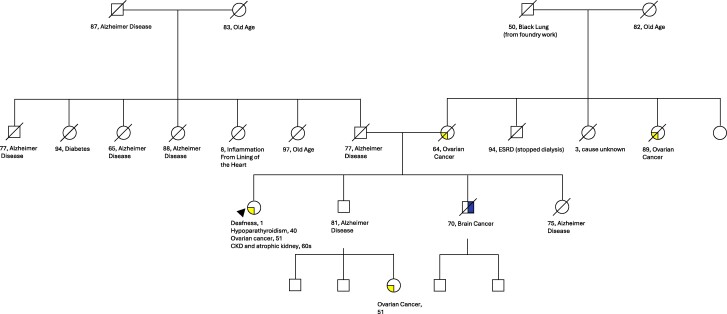
Hypoparathyroidism (hypoPTH), sensorineural deafness, and renal dysplasia (HDR) syndrome is a rare autosomal dominant condition with approximately 200 cases published. HDR syndrome is caused by variants of GATA binding protein 3 gene (GATA3), which encodes a transcription factor, with multiple types of GATA3 variants reported. We present the case of a 76-year-old woman who was diagnosed with hypoPTH when she was aged 40 years and transferred care to our institution. Further history elucidated presence of deafness at age 1 year and chronic kidney disease with a left atrophic kidney diagnosed in her 60 seconds. Genetic testing identified a novel GATA3 missense variant of unknown significance (c.791G > A, p.Cys264Tyr). There was no family history of hypoPTH, deafness, or renal disease, which might indicate incomplete penetrance or de novo mutation. Advanced modeling of protein sequence and biophysical properties predicts abnormal protein function, suggesting possible pathogenicity. In addition, a likely pathogenic variant in the same amino acid was previously described in a patient with HDR, supporting the in silico prediction of pathogenicity in our patient's variant. Syndromic hypoPTH should be considered in patients even if presenting later in life with presumed chronic isolated conditions. Genetic testing can guide further disease screening and family testing when appropriate.
甲状旁腺功能减退症(hypoPTH)、感音神经性耳聋和肾发育不良(HDR)综合征是一种罕见的常染色体显性遗传病,已发表病例约200例。HDR综合征是由编码转录因子的GATA结合蛋白3基因(GATA3)变异引起的,有多种类型的GATA3变异报道。我们提出的情况下,一个76岁的妇女谁被诊断为垂体功能低下时,她是40岁,并转移到我们的机构护理。进一步的病史表明,她在1岁时耳聋,并在60秒内诊断出慢性肾脏疾病伴左肾萎缩。基因检测鉴定出一种新的意义未知的GATA3错义变异(c.791G b> a, p.Cys264Tyr)。没有垂体功能低下、耳聋或肾脏疾病的家族史,这可能表明不完全外显或从头突变。蛋白质序列和生物物理特性的高级建模预测异常蛋白质功能,提示可能的致病性。此外,先前在一名HDR患者中发现了相同氨基酸的可能致病性变异,这支持了对该患者变异致病性的计算机预测。即使患者在晚年出现假定的慢性孤立性疾病,也应考虑综合征性甲状旁腺激素低下。基因检测可以在适当的时候指导进一步的疾病筛查和家庭检测。

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


