Peter T Nguyen, Simon G Coetzee, Irina Silacheva, Dennis J Hazelett
{"title":"Genome-wide association studies are enriched for interacting genes.","authors":"Peter T Nguyen, Simon G Coetzee, Irina Silacheva, Dennis J Hazelett","doi":"10.1186/s13040-024-00421-w","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>With recent advances in single cell technology, high-throughput methods provide unique insight into disease mechanisms and more importantly, cell type origin. Here, we used multi-omics data to understand how genetic variants from genome-wide association studies influence development of disease. We show in principle how to use genetic algorithms with normal, matching pairs of single-nucleus RNA- and ATAC-seq, genome annotations, and protein-protein interaction data to describe the genes and cell types collectively and their contribution to increased risk.</p><p><strong>Results: </strong>We used genetic algorithms to measure fitness of gene-cell set proposals against a series of objective functions that capture data and annotations. The highest information objective function captured protein-protein interactions. We observed significantly greater fitness scores and subgraph sizes in foreground vs. matching sets of control variants. Furthermore, our model reliably identified known targets and ligand-receptor pairs, consistent with prior studies.</p><p><strong>Conclusions: </strong>Our findings suggested that application of genetic algorithms to association studies can generate a coherent cellular model of risk from a set of susceptibility variants. Further, we showed, using breast cancer as an example, that such variants have a greater number of physical interactions than expected due to chance.</p>","PeriodicalId":48947,"journal":{"name":"Biodata Mining","volume":"18 1","pages":"3"},"PeriodicalIF":6.1000,"publicationDate":"2025-01-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11734473/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biodata Mining","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13040-024-00421-w","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
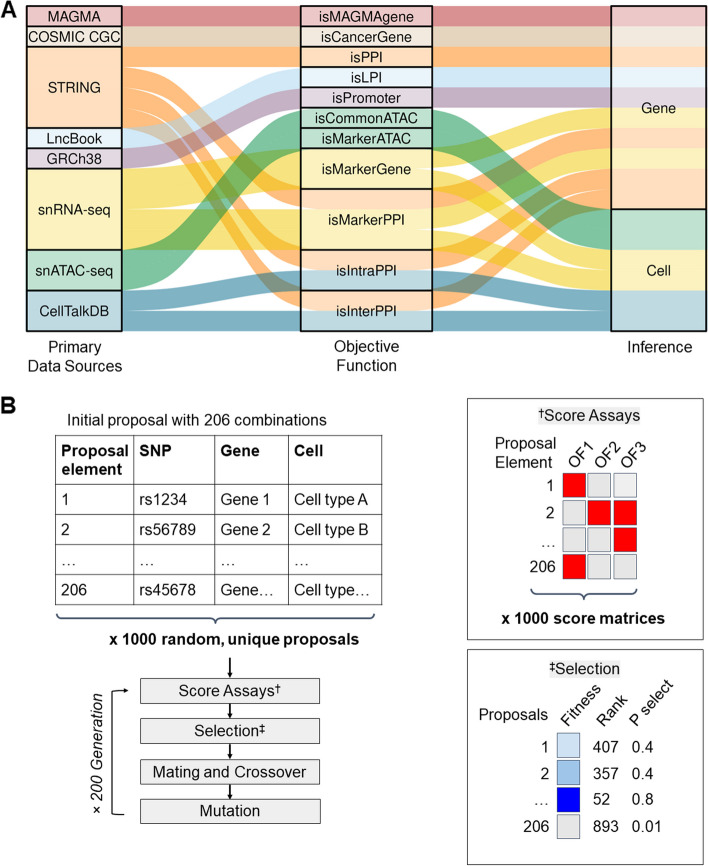
Background: With recent advances in single cell technology, high-throughput methods provide unique insight into disease mechanisms and more importantly, cell type origin. Here, we used multi-omics data to understand how genetic variants from genome-wide association studies influence development of disease. We show in principle how to use genetic algorithms with normal, matching pairs of single-nucleus RNA- and ATAC-seq, genome annotations, and protein-protein interaction data to describe the genes and cell types collectively and their contribution to increased risk.
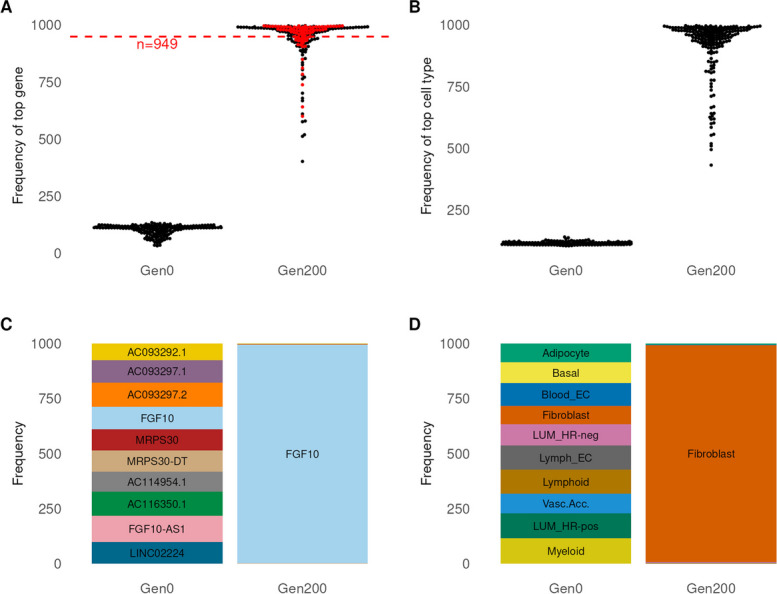
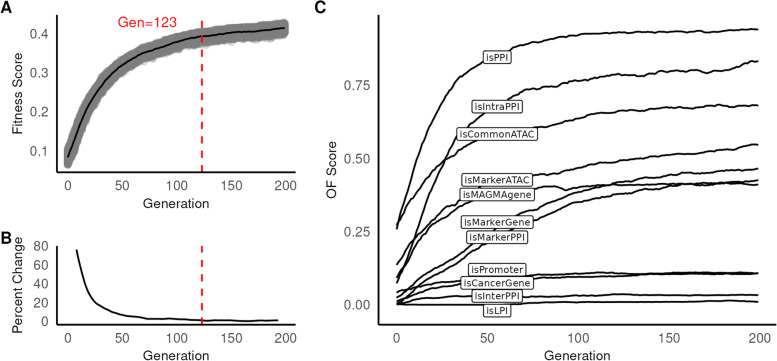
Results: We used genetic algorithms to measure fitness of gene-cell set proposals against a series of objective functions that capture data and annotations. The highest information objective function captured protein-protein interactions. We observed significantly greater fitness scores and subgraph sizes in foreground vs. matching sets of control variants. Furthermore, our model reliably identified known targets and ligand-receptor pairs, consistent with prior studies.
Conclusions: Our findings suggested that application of genetic algorithms to association studies can generate a coherent cellular model of risk from a set of susceptibility variants. Further, we showed, using breast cancer as an example, that such variants have a greater number of physical interactions than expected due to chance.
期刊介绍:
BioData Mining is an open access, open peer-reviewed journal encompassing research on all aspects of data mining applied to high-dimensional biological and biomedical data, focusing on computational aspects of knowledge discovery from large-scale genetic, transcriptomic, genomic, proteomic, and metabolomic data.
Topical areas include, but are not limited to:
-Development, evaluation, and application of novel data mining and machine learning algorithms.
-Adaptation, evaluation, and application of traditional data mining and machine learning algorithms.
-Open-source software for the application of data mining and machine learning algorithms.
-Design, development and integration of databases, software and web services for the storage, management, retrieval, and analysis of data from large scale studies.
-Pre-processing, post-processing, modeling, and interpretation of data mining and machine learning results for biological interpretation and knowledge discovery.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


