Aya Amer, Kathryn Murrell, Liza Edmonds, Isaac Bernhardt, Rhonda Akroyd, Bryony Ryder, Callum Wilson, Emma Glamuzina
{"title":"D,L-3-hydroxybutyrate in the treatment of glucose transporter 1 deficiency syndrome (Glut1DS)","authors":"Aya Amer, Kathryn Murrell, Liza Edmonds, Isaac Bernhardt, Rhonda Akroyd, Bryony Ryder, Callum Wilson, Emma Glamuzina","doi":"10.1002/jmd2.12461","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Background</h3>\n \n <p>Deficiency of the Glut1 transporter due to mono-allelic variants in <i>SLC2A1</i> causes hypoglycorrhachia, resulting in a neurological spectrum from neonatal epilepsy to adult-onset paroxysmal movement disorders (PMD). The brain utilises ketone bodies as an alternative energy source to glucose. Thus, early initiation of the ketogenic diet (KD) is standard care for Glut1 deficiency syndrome (Glut1DS). Commencement and adherence in older Glut1DS patients is difficult to achieve, leaving few treatment options. Oral D,L-3-hydroxybutyrate (D,L-3-HB) crosses the blood–brain barrier, making it a potential treatment for Glut1DS.</p>\n </section>\n \n <section>\n \n <h3> Methods</h3>\n \n <p>A retrospective case review of patients with Glut1DS under the Adult and Paediatric National Metabolic Service (APNMS) of New Zealand, treated with D,L-3-HB between 2012 and 2023 was performed. Clinical notes, standardised, neuropsychological assessments and subjective data on and off D,L-3-HB were obtained. The best on and off D,L-3-HB measures of working memory (WMI) and processing speed (PSI) were compared to assess the efficacy.</p>\n </section>\n \n <section>\n \n <h3> Results</h3>\n \n <p>D,L-3-HB was offered to 12 patients with Glut1DS (age 10–52 years). Compliance-dependent improvements in subjective, cognitive and adaptive function were reported by those who were reassessed on-treatment (9/12). Four reported improved PMD. Objective improvements were found in WM (9/9) and PS (6/9). Subjective improvements were reported in patients' health, wellbeing and independence.</p>\n </section>\n \n <section>\n \n <h3> Conclusions</h3>\n \n <p>KD remains standard of care for Glut1DS, but effective alternatives are lacking for those who do not tolerate this. D,L-3-HB was associated with improved WM, PS and perceived life quality in this small group of patients with Glut1DS, thus providing a potential treatment for this distinct group.</p>\n </section>\n </div>","PeriodicalId":14930,"journal":{"name":"JIMD reports","volume":"66 1","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2025-01-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11739118/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JIMD reports","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jmd2.12461","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 0
Abstract
Background
Deficiency of the Glut1 transporter due to mono-allelic variants in SLC2A1 causes hypoglycorrhachia, resulting in a neurological spectrum from neonatal epilepsy to adult-onset paroxysmal movement disorders (PMD). The brain utilises ketone bodies as an alternative energy source to glucose. Thus, early initiation of the ketogenic diet (KD) is standard care for Glut1 deficiency syndrome (Glut1DS). Commencement and adherence in older Glut1DS patients is difficult to achieve, leaving few treatment options. Oral D,L-3-hydroxybutyrate (D,L-3-HB) crosses the blood–brain barrier, making it a potential treatment for Glut1DS.
Methods
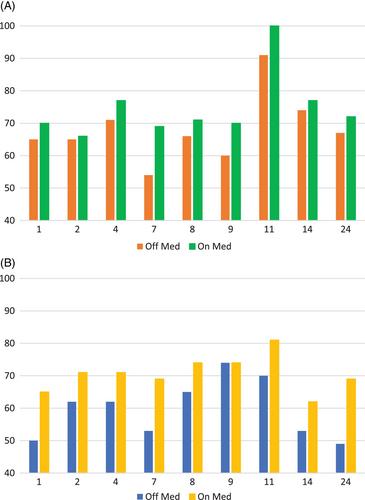
A retrospective case review of patients with Glut1DS under the Adult and Paediatric National Metabolic Service (APNMS) of New Zealand, treated with D,L-3-HB between 2012 and 2023 was performed. Clinical notes, standardised, neuropsychological assessments and subjective data on and off D,L-3-HB were obtained. The best on and off D,L-3-HB measures of working memory (WMI) and processing speed (PSI) were compared to assess the efficacy.
Results
D,L-3-HB was offered to 12 patients with Glut1DS (age 10–52 years). Compliance-dependent improvements in subjective, cognitive and adaptive function were reported by those who were reassessed on-treatment (9/12). Four reported improved PMD. Objective improvements were found in WM (9/9) and PS (6/9). Subjective improvements were reported in patients' health, wellbeing and independence.
Conclusions
KD remains standard of care for Glut1DS, but effective alternatives are lacking for those who do not tolerate this. D,L-3-HB was associated with improved WM, PS and perceived life quality in this small group of patients with Glut1DS, thus providing a potential treatment for this distinct group.
背景:SLC2A1的单等位基因变异导致Glut1转运体缺乏导致低糖血症,导致从新生儿癫痫到成人发作的阵发性运动障碍(PMD)的神经系统谱系。大脑利用酮体作为葡萄糖的替代能量来源。因此,早期开始生酮饮食(KD)是Glut1缺乏症(Glut1DS)的标准治疗。老年Glut1DS患者的开始和坚持治疗很难实现,留下很少的治疗选择。口服D, l -3-羟基丁酸酯(D,L-3-HB)可穿过血脑屏障,使其成为治疗谷氨酸变性的潜在药物。方法:回顾性分析新西兰成人和儿科国家代谢服务中心(APNMS) 2012年至2023年间接受D,L-3-HB治疗的Glut1DS患者的病例。获得临床记录、标准化的神经心理学评估和D、L-3-HB的主观数据。比较开、关D、L-3-HB工作记忆(WMI)和处理速度(PSI)的最佳测量值来评估疗效。结果:12例Glut1DS患者(年龄10-52岁)提供D,L-3-HB。治疗时重新评估的患者报告了主观、认知和适应功能的依从性改善(9/12)。四个报告了PMD的改进。WM(9/9)和PS(6/9)均有客观改善。据报告,患者的健康、幸福和独立性得到了主观改善。结论:KD仍然是谷胱甘肽缺乏症的标准治疗方案,但对于不能耐受的患者缺乏有效的替代方案。在这一小群Glut1DS患者中,D,L-3-HB与WM, PS和感知生活质量的改善有关,因此为这一独特的群体提供了潜在的治疗方法。

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


