{"title":"Mfgnn: Multi-Scale Feature-Attentive Graph Neural Networks for Molecular Property Prediction","authors":"Weiting Ye, Jingcheng Li, Xianfa Cai","doi":"10.1002/jcc.70011","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>In the realm of artificial intelligence-driven drug discovery (AIDD), accurately predicting the influence of molecular structures on their properties is a critical research focus. While deep learning models based on graph neural networks (GNNs) have made significant advancements in this area, prior studies have primarily concentrated on molecule-level representations, often neglecting the impact of functional group structures and the potential relationships between fragments on molecular property predictions. To address this gap, we introduce the multi-scale feature attention graph neural network (MfGNN), which enhances traditional atom-based molecular graph representations by incorporating fragment-level representations derived from chemically synthesizable BRICS fragments. MfGNN not only effectively captures both the structural information of molecules and the features of functional groups but also pays special attention to the potential relationships between fragments, exploring how they collectively influence molecular properties. This model integrates two core mechanisms: a graph attention mechanism that captures embeddings of molecules and functional groups, and a feature extraction module that systematically processes BRICS fragment-level features to uncover relationships among the fragments. Our comprehensive experiments demonstrate that MfGNN outperforms leading machine learning and deep learning models, achieving state-of-the-art performance in 8 out of 11 learning tasks across various domains, including physical chemistry, biophysics, physiology, and toxicology. Furthermore, ablation studies reveal that the integration of multi-scale feature information and the feature extraction module enhances the richness of molecular features, thereby improving the model's predictive capabilities.</p>\n </div>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 3","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2025-01-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70011","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
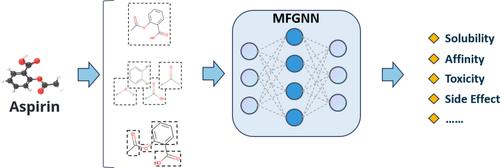
Abstract
In the realm of artificial intelligence-driven drug discovery (AIDD), accurately predicting the influence of molecular structures on their properties is a critical research focus. While deep learning models based on graph neural networks (GNNs) have made significant advancements in this area, prior studies have primarily concentrated on molecule-level representations, often neglecting the impact of functional group structures and the potential relationships between fragments on molecular property predictions. To address this gap, we introduce the multi-scale feature attention graph neural network (MfGNN), which enhances traditional atom-based molecular graph representations by incorporating fragment-level representations derived from chemically synthesizable BRICS fragments. MfGNN not only effectively captures both the structural information of molecules and the features of functional groups but also pays special attention to the potential relationships between fragments, exploring how they collectively influence molecular properties. This model integrates two core mechanisms: a graph attention mechanism that captures embeddings of molecules and functional groups, and a feature extraction module that systematically processes BRICS fragment-level features to uncover relationships among the fragments. Our comprehensive experiments demonstrate that MfGNN outperforms leading machine learning and deep learning models, achieving state-of-the-art performance in 8 out of 11 learning tasks across various domains, including physical chemistry, biophysics, physiology, and toxicology. Furthermore, ablation studies reveal that the integration of multi-scale feature information and the feature extraction module enhances the richness of molecular features, thereby improving the model's predictive capabilities.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


