Peter L. Rodríguez-Kessler and Alvaro Muñoz-Castro
{"title":"A Zn2-supported B7 wheel structure for the global minimum of the B7Zn2 cluster†","authors":"Peter L. Rodríguez-Kessler and Alvaro Muñoz-Castro","doi":"10.1039/D4CP04444D","DOIUrl":null,"url":null,"abstract":"<p >In this work, we employ density functional theory (DFT) to explore the structure of boron clusters doped with two zinc atoms (B<small><sub>7</sub></small>Zn<small><sub>2</sub></small> or Zn<small><sub>2</sub></small>B<small><sub>7</sub></small>). The results show that the most stable structure is a Zn<small><sub>2</sub></small> motif standing over a B<small><sub>7</sub></small> wheel, which is 0.89 eV lower in energy compared to the classical inverse-sandwich structure for B<small><sub>7</sub></small>TM<small><sub>2</sub></small> (TM = transition metal) clusters. The characteristics of these systems are evaluated by the IR spectra to guide plausible experimental realization. In addition, density of states, and bonding characteristics were evaluated. Our results denote the formation of an intermediate Zn–Zn bond order given by the key electron-acceptor nature of the B<small><sub>7</sub></small> motif, leading to a depopulation of antibonding Zn–Zn orbitals and population of the respective bonding orbitals. Thus, the evaluation and use of more electron-deficient supporting ligands may trigger a quest for the design of plausible structures featuring larger Zn–Zn elusive bond orders in stable species.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 7","pages":" 3583-3587"},"PeriodicalIF":2.9000,"publicationDate":"2025-01-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp04444d","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
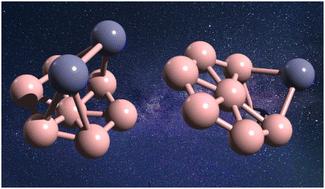
In this work, we employ density functional theory (DFT) to explore the structure of boron clusters doped with two zinc atoms (B7Zn2 or Zn2B7). The results show that the most stable structure is a Zn2 motif standing over a B7 wheel, which is 0.89 eV lower in energy compared to the classical inverse-sandwich structure for B7TM2 (TM = transition metal) clusters. The characteristics of these systems are evaluated by the IR spectra to guide plausible experimental realization. In addition, density of states, and bonding characteristics were evaluated. Our results denote the formation of an intermediate Zn–Zn bond order given by the key electron-acceptor nature of the B7 motif, leading to a depopulation of antibonding Zn–Zn orbitals and population of the respective bonding orbitals. Thus, the evaluation and use of more electron-deficient supporting ligands may trigger a quest for the design of plausible structures featuring larger Zn–Zn elusive bond orders in stable species.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


