Machine Learning Committee Neural Network Potential Energy Surfaces for Two-Dimensional Metal–Organic Frameworks
IF 3.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Two-dimensional (2D) layered metal–organic frameworks (MOFs) are gaining attention due to their unique structural and electronic properties with promising applications in compact electronic device fabrication. Long-time and large-scale molecular dynamics simulations of these materials can enhance and expedite the mapping out of their structure–property–function relationships for these applications. To make such simulations more feasible, herein, we construct a high-dimensional committee neural network potential (CNNP) for archetypal 2D MOFs Ni3(HIB)2 and Ni3(HITP)2 where HIB = hexaiminobenzene and HITP = hexaiminotriphenylene. We harness the power of active learning and committee neural networks to obtain a CNNP model by using only hundreds of snapshots from ab initio molecular dynamics (AIMD) trajectories. The developed CNNP model allows for simulations of thousands of atoms over extended time scales, which is typically unfeasible with AIMD simulations while maintaining the accuracy of the reference data. Our stable MD simulations based on the developed CNNP model reveal the flexible nature of the studied 2D MOFs at room temperature, including puckered layers, as opposed to the planar ones from 0 K electronic structure calculations. Furthermore, our model demonstrates transferability between bulk and monolayers, as well as different organic linkers. As the first model of its kind, we show that the high-dimensional CNNP models could be a reliable and effective approach for future studies on 2D MOFs.
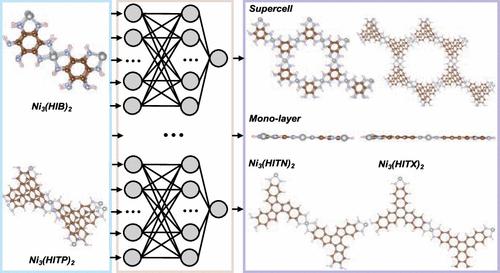
机器学习委员会二维金属-有机框架的神经网络势能面
二维(2D)层状金属有机框架(mof)由于其独特的结构和电子性能在紧凑型电子器件制造中具有广阔的应用前景而受到人们的关注。这些材料的长时间和大规模的分子动力学模拟可以增强和加快这些应用中它们的结构-性能-功能关系的绘制。为了使这种模拟更加可行,在此,我们为原型2D mof Ni3(HIB)2和Ni3(HITP)2构建了高维委员会神经网络电位(CNNP),其中HIB =己胺基苯,HITP =己胺基三苯。我们利用主动学习和委员会神经网络的力量,仅使用从头算分子动力学(AIMD)轨迹的数百个快照就获得了CNNP模型。开发的CNNP模型允许在延长的时间尺度上模拟数千个原子,这在保持参考数据准确性的情况下,在AIMD模拟中通常是不可实现的。我们基于开发的CNNP模型的稳定MD模拟揭示了所研究的二维mof在室温下的柔性性质,包括褶皱层,而不是从0 K电子结构计算的平面mof。此外,我们的模型证明了大块和单层之间以及不同有机连接物之间的可转移性。作为此类模型的第一个,我们表明高维CNNP模型可以为未来的二维mof研究提供可靠和有效的方法。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


