Decoding the Interplay of Hydrogen Bonding, Dispersion, and Steric Interactions in Conformational Isomerism Among Functionalized Pillar[n]arenes
IF 3.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
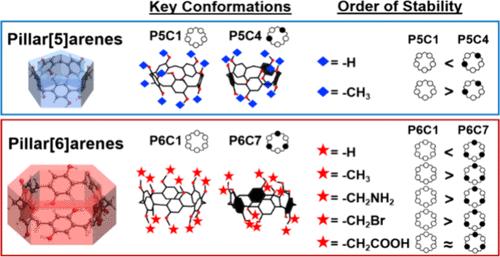
Pillar[n]arenes have garnered popularity due to their unique pillar-shaped structure, which results in hydrophobic cavities. These cavities facilitate the formation of inclusion complexes with guest molecules through noncovalent interactions such as π–π stacking, hydrogen bonding, and van der Waals interactions. Such host–guest interactions enable diverse functionalities in pillar[n]arenes, including molecule recognition, self-assembly, and encapsulation. Nevertheless, it is important to note that the host–guest properties of pillar[n]arenes can be influenced by conformational changes, primarily driven by the rotation of hydroquinone units about their methylene bridge axis. These structural changes can lead to variations in underlying noncovalent and steric interactions, impacting the overall stability of the host–guest system and potentially leading to selective uptake of guest molecules. Additionally, due to relative energy differences, we expect a distribution of pillar[n]arene conformations at thermal equilibrium. In this work, we employ density functional theory to evaluate ground-state electronic structures of pillar[n]arene conformations across pillar[n]arenes of various sizes and functionalizations. We have aimed to explore the impact of dispersion interactions, hydrogen bonding, and steric interactions on the overall energetics of pillar[n]arene conformations and determine the dominant conformation at 298 K using a Boltzmann-weighted distribution. The relative strengths of hydrogen bonds across various pillar[n]arene conformations have been examined using Bader’s quantum theory of atoms in molecules topological analysis. Furthermore, we also assessed the solvation of pillar[n]arenes in water using an implicit solvent model that unveils quantitative distinctions in hydrogen bonding and relative dispersion contributions among various pillar[n]arene conformations. Finally, pillar[n]arene conformations with more complex functional groups such as primary amine, alkyl bromide, and carboxylic acid have been studied to evaluate the interplay between underlying interactions such as hydrogen bonding, dispersion, and steric interactions and their collective impact on the structure and energetics of pillar[n]arene conformations.
功能化柱[n]芳烃构象异构中氢键、色散和空间相互作用的解码
柱[n]芳烃因其独特的柱状结构而广受欢迎,这种结构会产生疏水腔。这些空腔通过π -π堆叠、氢键和范德华相互作用等非共价相互作用促进与客体分子形成包合物。这种主客相互作用使柱[n]芳烃具有多种功能,包括分子识别、自组装和封装。然而,值得注意的是,柱[n]芳烃的主客体性质可能受到构象变化的影响,构象变化主要是由对苯二酚单位围绕其亚甲基桥轴的旋转驱动的。这些结构变化可能导致潜在的非共价和空间相互作用的变化,影响宿主-客体系统的整体稳定性,并可能导致客体分子的选择性摄取。此外,由于相对能量差异,我们预计在热平衡时柱[n]芳烃构象的分布。在这项工作中,我们采用密度泛函理论来评估不同尺寸和功能化的柱[n]芳烃构象的基态电子结构。我们的目的是探索分散相互作用、氢键和空间相互作用对柱[n]芳烃构象的整体能量学的影响,并利用玻尔兹曼加权分布确定298 K时的优势构象。在分子拓扑分析中,利用巴德的原子量子理论研究了不同柱[n]芳烃构象间氢键的相对强度。此外,我们还使用隐式溶剂模型评估了柱[n]芳烃在水中的溶剂化,该模型揭示了各种柱[n]芳烃构象之间氢键和相对分散贡献的定量差异。最后,对具有更复杂官能团(如伯胺、烷基溴和羧酸)的柱[n]芳烃构象进行了研究,以评估潜在相互作用(如氢键、分散和位向相互作用)之间的相互作用及其对柱[n]芳烃构象结构和能量学的共同影响。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


