{"title":"Improving Molecular Design with Direct Inverse Analysis of QSAR/QSPR Model.","authors":"Yuto Shino, Hiromasa Kaneko","doi":"10.1002/minf.202400227","DOIUrl":null,"url":null,"abstract":"<p><p>Recent advances in machine learning have significantly impacted molecular design, notably the molecular generation method combining the chemical variational autoencoder (VAE) with Gaussian mixture regression (GMR). In this method, a mathematical model is constructed with X as the latent variable of the molecule and Y as the target properties and activities. Through direct inverse analysis of this model, it is possible to generate molecules with the desired target properties. However, this approach outputs many strings that do not follow the simplified molecular input line entry system grammar and generates unrealistic chemical structures in which the properties and activity do not satisfy the target values. In this study, we focus on hierarchical VAE using molecular graphs to address these issues. We confirm that the combination of hierarchical VAE and GMR does not generate invalid outputs and returns molecules that simultaneously satisfy multiple target values. Moreover, we use this method to identify several molecules that are predicted to exhibit activity against drug targets.</p>","PeriodicalId":18853,"journal":{"name":"Molecular Informatics","volume":"44 1","pages":"e202400227"},"PeriodicalIF":3.1000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11724648/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Informatics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/minf.202400227","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
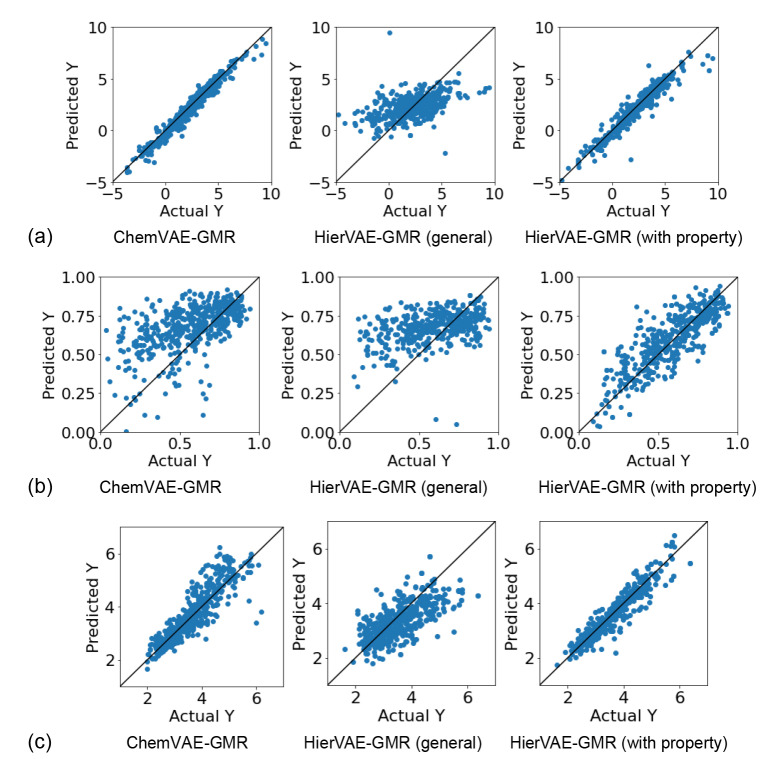
Recent advances in machine learning have significantly impacted molecular design, notably the molecular generation method combining the chemical variational autoencoder (VAE) with Gaussian mixture regression (GMR). In this method, a mathematical model is constructed with X as the latent variable of the molecule and Y as the target properties and activities. Through direct inverse analysis of this model, it is possible to generate molecules with the desired target properties. However, this approach outputs many strings that do not follow the simplified molecular input line entry system grammar and generates unrealistic chemical structures in which the properties and activity do not satisfy the target values. In this study, we focus on hierarchical VAE using molecular graphs to address these issues. We confirm that the combination of hierarchical VAE and GMR does not generate invalid outputs and returns molecules that simultaneously satisfy multiple target values. Moreover, we use this method to identify several molecules that are predicted to exhibit activity against drug targets.
期刊介绍:
Molecular Informatics is a peer-reviewed, international forum for publication of high-quality, interdisciplinary research on all molecular aspects of bio/cheminformatics and computer-assisted molecular design. Molecular Informatics succeeded QSAR & Combinatorial Science in 2010.
Molecular Informatics presents methodological innovations that will lead to a deeper understanding of ligand-receptor interactions, macromolecular complexes, molecular networks, design concepts and processes that demonstrate how ideas and design concepts lead to molecules with a desired structure or function, preferably including experimental validation.
The journal''s scope includes but is not limited to the fields of drug discovery and chemical biology, protein and nucleic acid engineering and design, the design of nanomolecular structures, strategies for modeling of macromolecular assemblies, molecular networks and systems, pharmaco- and chemogenomics, computer-assisted screening strategies, as well as novel technologies for the de novo design of biologically active molecules. As a unique feature Molecular Informatics publishes so-called "Methods Corner" review-type articles which feature important technological concepts and advances within the scope of the journal.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


