{"title":"Identification of a novel missense variant in the <i>AVP</i> gene in a Japanese pedigree with familial neurohypophyseal diabetes insipidus.","authors":"Daiei Kojima, Masami Shibata, Hiroaki Shikano, Yoshihiro Maruo, Hidehiko Fujii","doi":"10.1297/cpe.2024-0067","DOIUrl":null,"url":null,"abstract":"<p><p>Familial neurohypophyseal diabetes insipidus is a rare genetic disease caused by <i>AVP</i> gene variants and is characterized by progressive polyuria and polydipsia in early childhood. Herein, we have reported the clinical symptoms and genetic test results of a Japanese patient with a family history of polyuria and polydipsia for over five generations. The proband was a 6-yr-old boy who was referred for the evaluation of polyuria and polydipsia. A hypertonic saline infusion test showed no increase in AVP levels and a water deprivation test followed by vasopressin administration confirmed the diagnosis of central diabetes insipidus. Genetic analyses of the patient and his affected mother revealed a novel heterozygous missense variant (c.308T>A, p.V103D). This variant was located in the region encoding the neurophysin II moiety. Computational analysis predicted that p.V103D is pathogenic, and a structural change was detected by viewing the three-dimensional structure of the protein model. To our knowledge, this is the first study to identify a novel missense variant, p.V103D, in a Japanese family with central diabetes insipidus. These findings expand the panel of <i>AVP</i> variants and facilitate the genetic diagnosis of familial neurohypophyseal diabetes insipidus.</p>","PeriodicalId":10678,"journal":{"name":"Clinical Pediatric Endocrinology","volume":"34 1","pages":"77-82"},"PeriodicalIF":1.2000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11701011/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Pediatric Endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1297/cpe.2024-0067","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/30 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
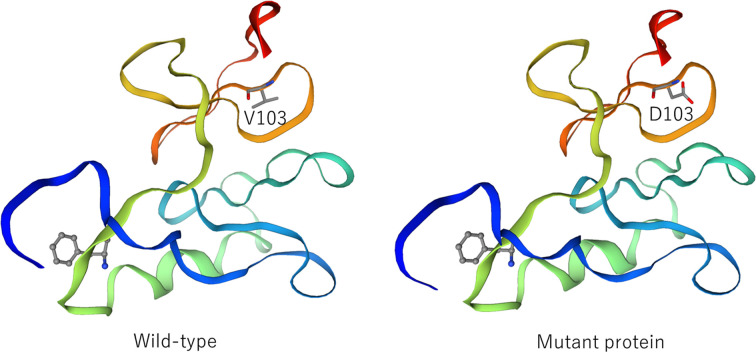
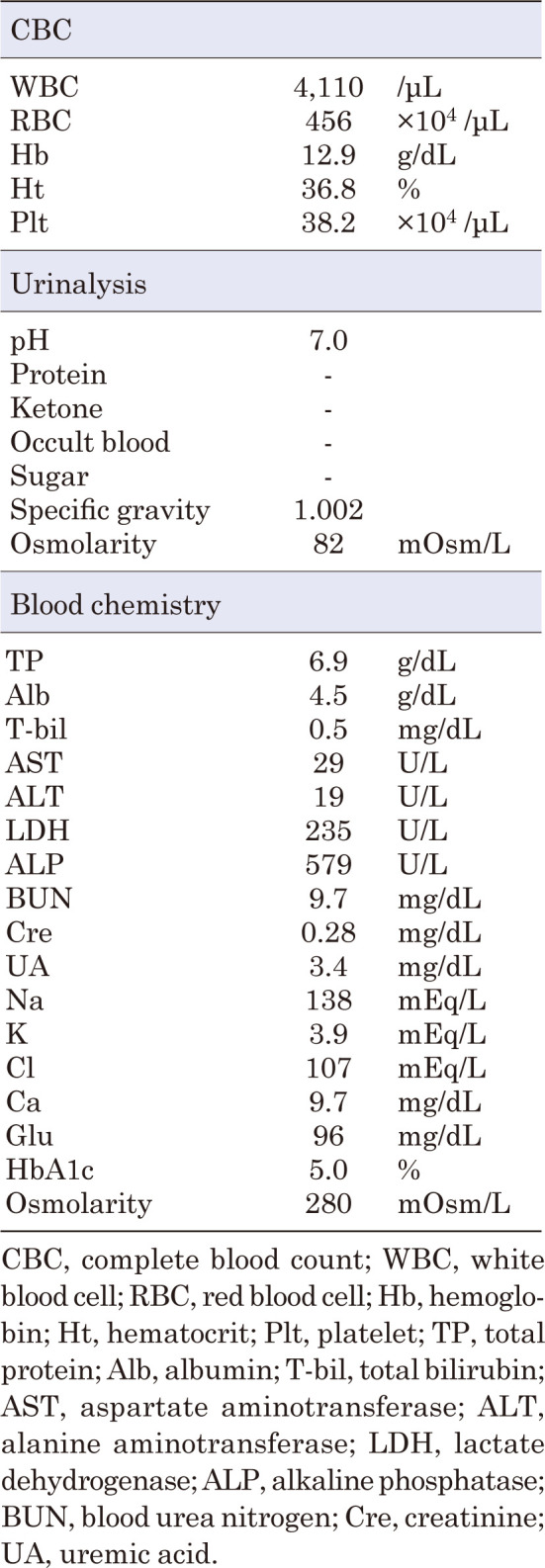
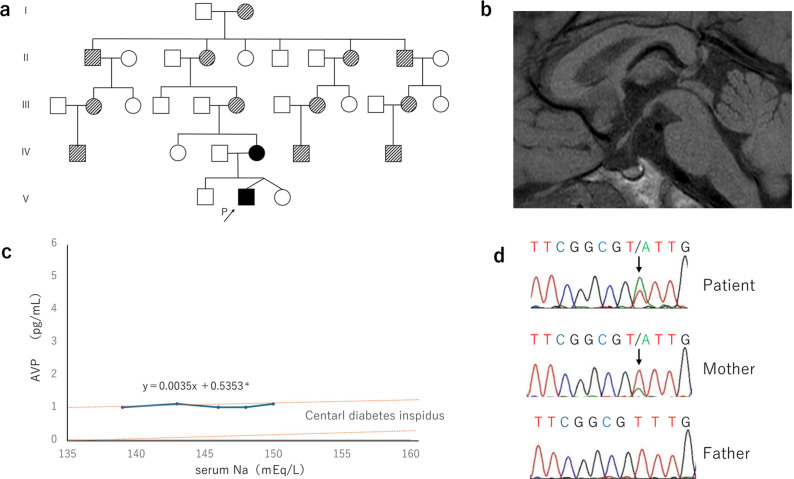
Familial neurohypophyseal diabetes insipidus is a rare genetic disease caused by AVP gene variants and is characterized by progressive polyuria and polydipsia in early childhood. Herein, we have reported the clinical symptoms and genetic test results of a Japanese patient with a family history of polyuria and polydipsia for over five generations. The proband was a 6-yr-old boy who was referred for the evaluation of polyuria and polydipsia. A hypertonic saline infusion test showed no increase in AVP levels and a water deprivation test followed by vasopressin administration confirmed the diagnosis of central diabetes insipidus. Genetic analyses of the patient and his affected mother revealed a novel heterozygous missense variant (c.308T>A, p.V103D). This variant was located in the region encoding the neurophysin II moiety. Computational analysis predicted that p.V103D is pathogenic, and a structural change was detected by viewing the three-dimensional structure of the protein model. To our knowledge, this is the first study to identify a novel missense variant, p.V103D, in a Japanese family with central diabetes insipidus. These findings expand the panel of AVP variants and facilitate the genetic diagnosis of familial neurohypophyseal diabetes insipidus.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


