Deep Proteome Coverage of Microglia Using a Streamlined Data-Independent Acquisition-Based Proteomic Workflow: Method Consideration for a Phenotypically Diverse Cell Type.
Jessica Wohlfahrt, Jennifer Guergues, Stanley M Stevens
{"title":"Deep Proteome Coverage of Microglia Using a Streamlined Data-Independent Acquisition-Based Proteomic Workflow: Method Consideration for a Phenotypically Diverse Cell Type.","authors":"Jessica Wohlfahrt, Jennifer Guergues, Stanley M Stevens","doi":"10.3390/proteomes12040035","DOIUrl":null,"url":null,"abstract":"<p><p>As the primary innate immune cells of the brain, microglia play a key role in various homeostatic and disease-related processes. To carry out their numerous functions, microglia adopt a wide range of phenotypic states. The proteomic landscape represents a more accurate molecular representation of these phenotypes; however, microglia present unique challenges for proteomic analysis. This study implemented a streamlined liquid- and gas-phase fractionation method with data-dependent acquisition (DDA) and parallel accumulation-serial fragmentation (PASEF) analysis on a TIMS-TOF instrument to compile a comprehensive protein library obtained from adult-derived, immortalized mouse microglia with low starting material (10 µg). The empirical library consisted of 9140 microglial proteins and was utilized to identify an average of 7264 proteins/run from single-shot, data-independent acquisition (DIA)-based analysis microglial cell lysate digest (200 ng). Additionally, a predicted library facilitated the identification of 7519 average proteins/run from the same DIA data, revealing complementary coverage compared with the empirical library and collectively increasing coverage to approximately 8000 proteins. Importantly, several microglia-relevant pathways were uniquely identified with the empirical library approach. Overall, we report a simplified, reproducible approach to address the proteome complexity of microglia using low sample input and show the importance of library optimization for this phenotypically diverse cell type.</p>","PeriodicalId":20877,"journal":{"name":"Proteomes","volume":"12 4","pages":""},"PeriodicalIF":3.6000,"publicationDate":"2024-11-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11679481/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Proteomes","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/proteomes12040035","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
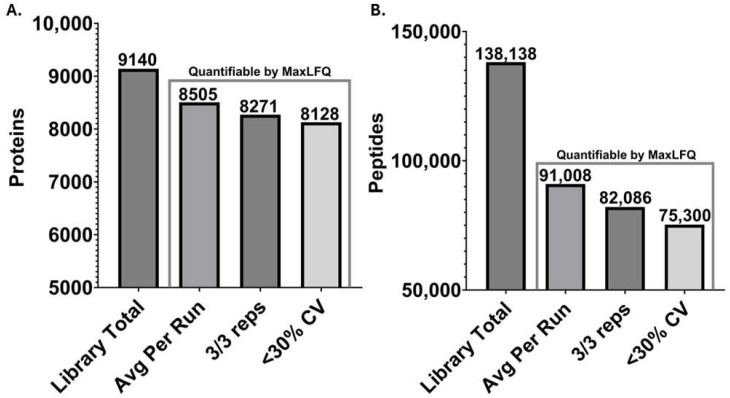
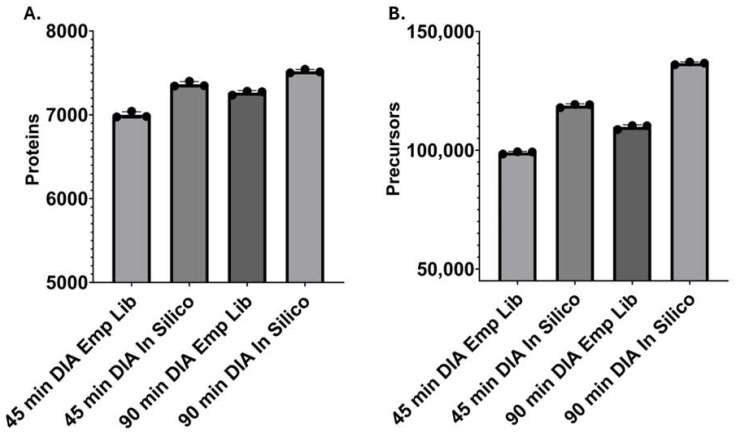
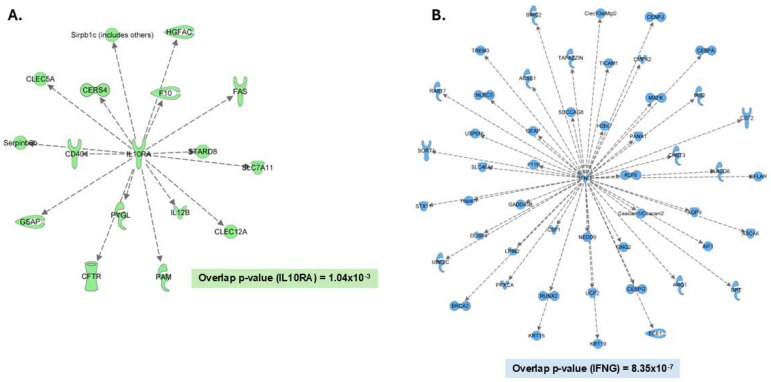
As the primary innate immune cells of the brain, microglia play a key role in various homeostatic and disease-related processes. To carry out their numerous functions, microglia adopt a wide range of phenotypic states. The proteomic landscape represents a more accurate molecular representation of these phenotypes; however, microglia present unique challenges for proteomic analysis. This study implemented a streamlined liquid- and gas-phase fractionation method with data-dependent acquisition (DDA) and parallel accumulation-serial fragmentation (PASEF) analysis on a TIMS-TOF instrument to compile a comprehensive protein library obtained from adult-derived, immortalized mouse microglia with low starting material (10 µg). The empirical library consisted of 9140 microglial proteins and was utilized to identify an average of 7264 proteins/run from single-shot, data-independent acquisition (DIA)-based analysis microglial cell lysate digest (200 ng). Additionally, a predicted library facilitated the identification of 7519 average proteins/run from the same DIA data, revealing complementary coverage compared with the empirical library and collectively increasing coverage to approximately 8000 proteins. Importantly, several microglia-relevant pathways were uniquely identified with the empirical library approach. Overall, we report a simplified, reproducible approach to address the proteome complexity of microglia using low sample input and show the importance of library optimization for this phenotypically diverse cell type.
ProteomesBiochemistry, Genetics and Molecular Biology-Clinical Biochemistry
CiteScore
6.50
自引率
3.00%
发文量
37
审稿时长
11 weeks
期刊介绍:
Proteomes (ISSN 2227-7382) is an open access, peer reviewed journal on all aspects of proteome science. Proteomes covers the multi-disciplinary topics of structural and functional biology, protein chemistry, cell biology, methodology used for protein analysis, including mass spectrometry, protein arrays, bioinformatics, HTS assays, etc. Our aim is to encourage scientists to publish their experimental and theoretical results in as much detail as possible. Therefore, there is no restriction on the length of papers. Scope: -whole proteome analysis of any organism -disease/pharmaceutical studies -comparative proteomics -protein-ligand/protein interactions -structure/functional proteomics -gene expression -methodology -bioinformatics -applications of proteomics

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


