Quantitatively Connecting Experimental Time–Temperature–Superposition–Breakdown of Polymers near the Glass Transition to Dynamic Heterogeneity Via the Heterogeneous Rouse Model
IF 5.1
1区 化学
Q1 POLYMER SCIENCE
引用次数: 0
Abstract
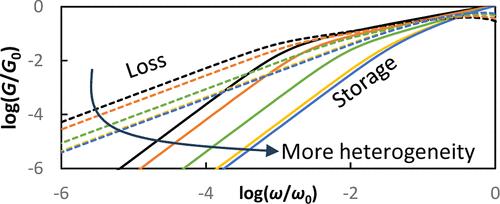
Polymers near the glass transition temperature Tg often exhibit a breakdown of time–temperature–superposition (TTS), with chain relaxation times and viscosity exhibiting a weaker temperature dependence than segmental relaxation times. The origin of this onset of thermorheological complexity has remained unsettled and a matter of debate. Here we extend the Heterogeneous Rouse Model (HRM), which generalizes the Rouse model to account for dynamic heterogeneity, to make predictions for the relaxation modulus G(t) and complex modulus G*(ω) of unentangled polymers near Tg. The HRM predicts that G(t) and G*(ω) exhibit enhanced effective scaling exponents in the Rouse regime in the presence of dynamic heterogeneity, with a more rapid decay from the glassy plateau emerging as the system becomes more dynamically heterogeneous on cooling. This behavior is predicted to emerge from a strand-length dependence of the moment of the segmental mobility distribution probed by chain dynamics. We show that the HRM predictions are in good accord with experimental complex modulus data for polystyrene, poly(methyl methacrylate), and poly(2-vinylpyridine). The HRM also predicts the onset of distinct temperature dependences among chain scale quantities such as terminal relaxation time and viscosity in our experimental systems, apparently resolving one of the most significant standing objections to a heterogeneity-based origin of TTS-breakdown. The HRM thus provides a generalized theory of the chain-scale linear rheological response of unentangled polymers near Tg, accounting for the origin of TTS-breakdown at a molecular mechanistic level. It also points toward a new strategy of inferring the dynamic heterogeneity of glass-forming polymeric systems from the temperature–evolution of modified scaling in the Rouse regime.
通过非均相Rouse模型定量地将实验时间-温度-叠加-接近玻璃化转变的聚合物击穿与动态非均质联系起来
接近玻璃化转变温度Tg的聚合物通常表现出时间-温度-叠加(TTS)的破裂,链弛豫时间和粘度表现出比段弛豫时间更弱的温度依赖性。这种热流变复杂性的起源仍然没有解决和争论的问题。在这里,我们扩展了异构Rouse模型(HRM),它将Rouse模型推广到考虑动态非均质性,以预测未纠缠聚合物在Tg附近的松弛模量G(t)和复模量G*(ω)。HRM预测,在动态非均质存在的情况下,G(t)和G*(ω)在劳斯体系中表现出增强的有效标度指数,随着系统在冷却时变得更加动态非均质,从玻璃状平台出现更快的衰减。这种行为被预测来自链动力学探测的节段迁移率分布的矩的链长依赖。研究表明,HRM预测与聚苯乙烯、聚甲基丙烯酸甲酯和聚2-乙烯基吡啶的实验复模量数据吻合良好。HRM还预测了链尺度量(如终端松弛时间和实验系统中的粘度)之间不同温度依赖性的开始,显然解决了对基于异质性的tts分解起源的最重要的反对意见之一。因此,HRM提供了Tg附近未纠缠聚合物的链尺度线性流变响应的广义理论,在分子机制水平上解释了tts击穿的起源。它还指出了一种新的策略来推断玻璃形成聚合物体系的动态非均质性,从温度演化的修正尺度在劳斯制度。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Macromolecules
工程技术-高分子科学
CiteScore
9.30
自引率
16.40%
发文量
942
审稿时长
2 months
期刊介绍:
Macromolecules publishes original, fundamental, and impactful research on all aspects of polymer science. Topics of interest include synthesis (e.g., controlled polymerizations, polymerization catalysis, post polymerization modification, new monomer structures and polymer architectures, and polymerization mechanisms/kinetics analysis); phase behavior, thermodynamics, dynamic, and ordering/disordering phenomena (e.g., self-assembly, gelation, crystallization, solution/melt/solid-state characteristics); structure and properties (e.g., mechanical and rheological properties, surface/interfacial characteristics, electronic and transport properties); new state of the art characterization (e.g., spectroscopy, scattering, microscopy, rheology), simulation (e.g., Monte Carlo, molecular dynamics, multi-scale/coarse-grained modeling), and theoretical methods. Renewable/sustainable polymers, polymer networks, responsive polymers, electro-, magneto- and opto-active macromolecules, inorganic polymers, charge-transporting polymers (ion-containing, semiconducting, and conducting), nanostructured polymers, and polymer composites are also of interest. Typical papers published in Macromolecules showcase important and innovative concepts, experimental methods/observations, and theoretical/computational approaches that demonstrate a fundamental advance in the understanding of polymers.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


