Anna Kasprzyk-Pawelec, Mingjun Tan, Raneen Rahhal, Alec McIntosh, Harvey R. Fernandez, Rami M. Mosaoa, Lei Jiang, Gray W. Pearson, Eric Glasgow, Jerry Vockley, Christopher Albanese, Maria Laura Avantaggiati
{"title":"Inactivation of the SLC25A1 gene during embryogenesis induces a unique senescence program controlled by p53","authors":"Anna Kasprzyk-Pawelec, Mingjun Tan, Raneen Rahhal, Alec McIntosh, Harvey R. Fernandez, Rami M. Mosaoa, Lei Jiang, Gray W. Pearson, Eric Glasgow, Jerry Vockley, Christopher Albanese, Maria Laura Avantaggiati","doi":"10.1038/s41418-024-01428-w","DOIUrl":null,"url":null,"abstract":"<p>Germline inactivating mutations of the <i>SLC25A1</i> gene contribute to various human disorders, including Velocardiofacial (VCFS), DiGeorge (DGS) syndromes and combined D/L-2-hydroxyglutaric aciduria (D/L-2HGA), a severe systemic disease characterized by the accumulation of 2-hydroxyglutaric acid (2HG). The mechanisms by which <i>SLC25A1</i> loss leads to these syndromes remain largely unclear. Here, we describe a mouse model of <i>SLC25A1</i> deficiency that mimics human VCFS/DGS and D/L-2HGA. Surprisingly, inactivation of both <i>Slc25a1</i> alleles results in alterations in the development of multiple organs, and in a severe proliferation defect by activating two senescence programs, oncogene-induced senescence (OIS) and mitochondrial dysfunction-induced senescence (MiDAS), which converge upon the induction of the p53 tumor suppressor. Mechanistically, cells and tissues with dysfunctional SLC25A1 protein undergo metabolic and transcriptional rewiring leading to the accumulation of 2HG <i>via</i> a non-canonical pathway and to the depletion of nicotinamide adenine dinucleotide, NAD<sup>+</sup>, which trigger senescence. Replenishing the pool of NAD<sup>+</sup> or promoting the clearance of 2HG rescues the proliferation defect of cells with dysfunctional SLC25A1 in a cooperative fashion. Further, removal of p53 activity <i>via</i> RNA interference restores proliferation, indicating that p53 acts as a critical barrier to the expansion of cells lacking functional SLC25A1. These findings reveal unexpected pathogenic roles of senescence and of p53 in D/L-2HGA and identify potential therapeutic strategies to correct salient molecular alterations driving this disease.</p>","PeriodicalId":9731,"journal":{"name":"Cell Death and Differentiation","volume":"27 1","pages":""},"PeriodicalIF":13.7000,"publicationDate":"2024-12-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell Death and Differentiation","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1038/s41418-024-01428-w","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
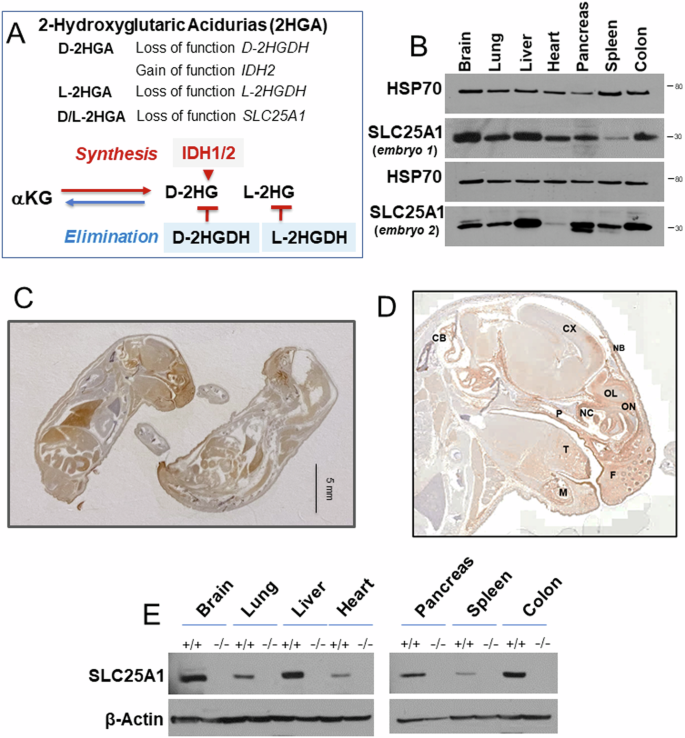
Germline inactivating mutations of the SLC25A1 gene contribute to various human disorders, including Velocardiofacial (VCFS), DiGeorge (DGS) syndromes and combined D/L-2-hydroxyglutaric aciduria (D/L-2HGA), a severe systemic disease characterized by the accumulation of 2-hydroxyglutaric acid (2HG). The mechanisms by which SLC25A1 loss leads to these syndromes remain largely unclear. Here, we describe a mouse model of SLC25A1 deficiency that mimics human VCFS/DGS and D/L-2HGA. Surprisingly, inactivation of both Slc25a1 alleles results in alterations in the development of multiple organs, and in a severe proliferation defect by activating two senescence programs, oncogene-induced senescence (OIS) and mitochondrial dysfunction-induced senescence (MiDAS), which converge upon the induction of the p53 tumor suppressor. Mechanistically, cells and tissues with dysfunctional SLC25A1 protein undergo metabolic and transcriptional rewiring leading to the accumulation of 2HG via a non-canonical pathway and to the depletion of nicotinamide adenine dinucleotide, NAD+, which trigger senescence. Replenishing the pool of NAD+ or promoting the clearance of 2HG rescues the proliferation defect of cells with dysfunctional SLC25A1 in a cooperative fashion. Further, removal of p53 activity via RNA interference restores proliferation, indicating that p53 acts as a critical barrier to the expansion of cells lacking functional SLC25A1. These findings reveal unexpected pathogenic roles of senescence and of p53 in D/L-2HGA and identify potential therapeutic strategies to correct salient molecular alterations driving this disease.
SLC25A1基因的种系失活突变可导致多种人类疾病,包括速心面(VCFS)、DiGeorge (DGS)综合征和合并D/ l -2-羟基戊二酸尿症(D/L-2HGA),后者是一种以2-羟基戊二酸(2HG)积累为特征的严重全身性疾病。SLC25A1缺失导致这些综合征的机制在很大程度上仍不清楚。在这里,我们描述了一个模拟人类VCFS/DGS和D/L-2HGA的SLC25A1缺陷小鼠模型。令人惊讶的是,Slc25a1等位基因的失活会导致多个器官发育的改变,并通过激活两个衰老程序导致严重的增殖缺陷,癌基因诱导衰老(OIS)和线粒体功能障碍诱导衰老(MiDAS),这两个衰老程序汇聚在p53肿瘤抑制因子的诱导上。从机制上说,SLC25A1蛋白功能失调的细胞和组织通过代谢和转录重新布线,通过非规范途径导致2HG的积累,并导致引发衰老的烟酰胺腺嘌呤二核苷酸(NAD+)的消耗。补充NAD+池或促进2HG的清除以一种合作的方式拯救SLC25A1功能失调的细胞的增殖缺陷。此外,通过RNA干扰去除p53活性可以恢复增殖,这表明p53是缺乏功能SLC25A1的细胞增殖的关键屏障。这些发现揭示了衰老和p53在D/L-2HGA中意想不到的致病作用,并确定了纠正导致该疾病的显着分子改变的潜在治疗策略。
期刊介绍:
Mission, vision and values of Cell Death & Differentiation:
To devote itself to scientific excellence in the field of cell biology, molecular biology, and biochemistry of cell death and disease.
To provide a unified forum for scientists and clinical researchers
It is committed to the rapid publication of high quality original papers relating to these subjects, together with topical, usually solicited, reviews, meeting reports, editorial correspondence and occasional commentaries on controversial and scientifically informative issues.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


