Modulating the Electronic Properties of Orthorhombic Mo2C Surfaces with Strain and Defects: Insights from First-Principles Calculations
IF 3.3
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
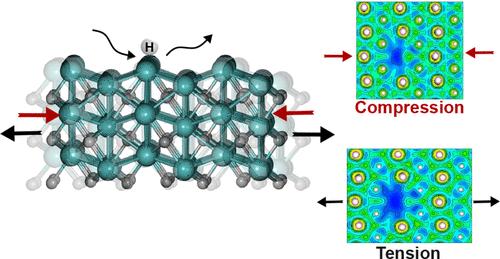
Mo2C is an efficient and cost-effective catalyst for hydrogenation reactions that are crucial for chemical synthesis and renewable energy applications. In this study, we investigate the electronic and adsorption properties of orthorhombic Mo2C (001) with three different surface terminations using density functional theory. By introducing Mo and C vacancies and substituting Mo with Ti, we evaluate the effect of defects on the electron localization function (ELF), projected density of states, and hydrogen adsorption behavior. The results show that Mo atom vacancies significantly disrupt the ELF distribution, while C atom vacancies and Ti substitutions have little effect. Tensile or compressive strain applied to the surfaces modulates the ELF for surfaces with Mo defects but has little effect on systems with C vacancies or Ti substitutions. We also examine how defects and strain affect hydrogen adsorption on the Mo2C surfaces to understand the potential effect on catalytic performance. The findings of this study highlight the importance of defect and strain conditions in the catalytic efficiency of orthorhombic Mo2C and offer valuable insight into designing strain- and defect-engineered catalysts with enhanced hydrogen adsorption and desorption properties, paving the way for more efficient and selective hydrogenation processes.
带应变和缺陷的正交Mo2C表面的电子特性调制:来自第一性原理计算的见解
Mo2C是一种高效、经济的氢化反应催化剂,对化学合成和可再生能源应用至关重要。在本研究中,我们利用密度泛函理论研究了具有三种不同表面末端的正交Mo2C(001)的电子和吸附性质。通过引入Mo和C空位,并用Ti取代Mo,我们评估了缺陷对电子定位函数(ELF)、投射态密度和氢吸附行为的影响。结果表明,Mo原子空位明显破坏了极低频分布,而C原子空位和Ti取代对极低频分布影响不大。施加于表面的拉伸或压缩应变可调节含有Mo缺陷表面的极低频光谱,但对含有C空位或Ti取代的体系影响不大。我们还研究了缺陷和应变如何影响Mo2C表面的氢吸附,以了解对催化性能的潜在影响。本研究结果强调了缺陷和应变条件对正交Mo2C催化效率的重要性,并为设计具有增强氢吸附和解吸性能的应变和缺陷工程催化剂提供了有价值的见解,为更高效和选择性的加氢工艺铺平了道路。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


