Statistical, Bottom-Up Model for Chemical Diffusion Based on Atomic Vacancy Sublattice Configurations in Layered Lithium Nickel Oxide Cathode Materials
Stéphane B. Olou'ou Guifo, Jonathan E. Mueller, Torsten Markus
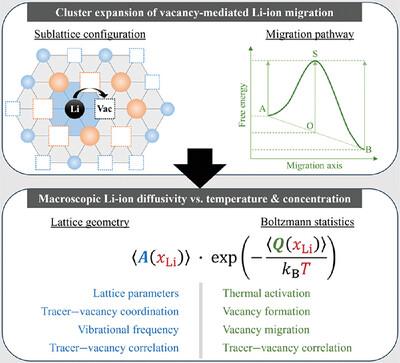
{"title":"Statistical, Bottom-Up Model for Chemical Diffusion Based on Atomic Vacancy Sublattice Configurations in Layered Lithium Nickel Oxide Cathode Materials","authors":"Stéphane B. Olou'ou Guifo, Jonathan E. Mueller, Torsten Markus","doi":"10.1002/adts.202400917","DOIUrl":null,"url":null,"abstract":"<p>To investigate the influence of the local environment on Li-ion diffusivity in layered lithium nickel oxide (Li<sub><i>x</i></sub>NiO<sub>2</sub>) cathodes, a bottom-up, multiscale-modeling approach is applied, utilizing density functional theory (DFT) with corrected Coulomb and van der Waals interactions to describe the energy-structure relationship of Li<sub><i>x</i></sub>NiO<sub>2</sub> (<i>x</i> = 0 – 1) in good agreement with previous experiments. The UNiversal CLuster Expansion (UNCLE) is employed to construct high-probability Li–vacancy configurations and the Nudged Elastic Band (NEB) method to compute energy barriers for representative Li diffusion mechanisms. By fitting a cluster expansion model to these barriers, diffusion barriers are determined for all possible Li–vacancy configurations within a nearest-neighbor approximation. Based on this description, Li-concentration-dependent diffusion coefficients are predicted for the entire Li-concentration range. For the Li<sub><i>x</i></sub>NiO<sub>2</sub> crystal lattice, the computed Li chemical diffusivities well lie within experimental ranges, namely 10<sup>−11</sup> – 10<sup>−8</sup> cm<sup>2</sup> s<sup>−1</sup>, at room temperature with activation energies around 37.9 kJ mol<sup>−1</sup>. The maximum diffusivity of 4.23 × 10<sup>−8</sup> cm<sup>2</sup> s<sup>−1</sup> is identified at <i>x</i> = 0.63. The new analytical, self-consistent approach here relies on configurational samplings of individual atomistic mechanisms and can be applied to investigate diffusion properties in further dilute and concentrated alloy systems more efficiently than common numerical procedures.</p>","PeriodicalId":7219,"journal":{"name":"Advanced Theory and Simulations","volume":"8 4","pages":""},"PeriodicalIF":2.9000,"publicationDate":"2024-12-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Advanced Theory and Simulations","FirstCategoryId":"5","ListUrlMain":"https://advanced.onlinelibrary.wiley.com/doi/10.1002/adts.202400917","RegionNum":4,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
To investigate the influence of the local environment on Li-ion diffusivity in layered lithium nickel oxide (LixNiO2) cathodes, a bottom-up, multiscale-modeling approach is applied, utilizing density functional theory (DFT) with corrected Coulomb and van der Waals interactions to describe the energy-structure relationship of LixNiO2 (x = 0 – 1) in good agreement with previous experiments. The UNiversal CLuster Expansion (UNCLE) is employed to construct high-probability Li–vacancy configurations and the Nudged Elastic Band (NEB) method to compute energy barriers for representative Li diffusion mechanisms. By fitting a cluster expansion model to these barriers, diffusion barriers are determined for all possible Li–vacancy configurations within a nearest-neighbor approximation. Based on this description, Li-concentration-dependent diffusion coefficients are predicted for the entire Li-concentration range. For the LixNiO2 crystal lattice, the computed Li chemical diffusivities well lie within experimental ranges, namely 10−11 – 10−8 cm2 s−1, at room temperature with activation energies around 37.9 kJ mol−1. The maximum diffusivity of 4.23 × 10−8 cm2 s−1 is identified at x = 0.63. The new analytical, self-consistent approach here relies on configurational samplings of individual atomistic mechanisms and can be applied to investigate diffusion properties in further dilute and concentrated alloy systems more efficiently than common numerical procedures.
期刊介绍:
Advanced Theory and Simulations is an interdisciplinary, international, English-language journal that publishes high-quality scientific results focusing on the development and application of theoretical methods, modeling and simulation approaches in all natural science and medicine areas, including:
materials, chemistry, condensed matter physics
engineering, energy
life science, biology, medicine
atmospheric/environmental science, climate science
planetary science, astronomy, cosmology
method development, numerical methods, statistics






 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


