Elsa Balduzzi, Wenlu Yin, Jean-Christophe Lambry, Hannu Myllykallio, Alexey Aleksandrov
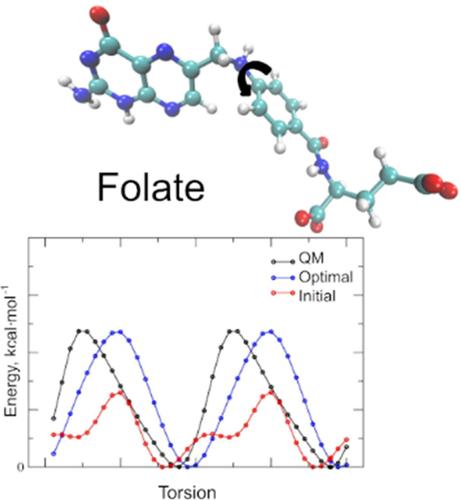
{"title":"Additive CHARMM Force Field for Pterins and Folates","authors":"Elsa Balduzzi, Wenlu Yin, Jean-Christophe Lambry, Hannu Myllykallio, Alexey Aleksandrov","doi":"10.1002/jcc.27548","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>Folates comprise a crucial class of biologically active compounds related to folic acid, playing a vital role in numerous enzymatic reactions. One-carbon metabolism, facilitated by the folate cofactor, supports numerous physiological processes, including biosynthesis, amino acid homeostasis, epigenetic maintenance, and redox defense. Folates share a common pterin heterocyclic ring structure capable of undergoing redox reactions and existing in various protonation states. This study aimed to derive molecular mechanics (MM) parameters compatible with the CHARMM36 all-atom additive force field for pterins and biologically important folates, including pterin, biopterin, and folic acid. Three redox forms were considered: oxidized, dihydrofolate, and tetrahydrofolate states. Across all protonation states, a total of 18 folates were parameterized. Partial charges were derived using the CHARMM force field parametrization protocol, based on targeting reference quantum mechanics monohydrate interactions, electrostatic potential, and dipole moment. Bonded terms were parameterized using one-dimensional adiabatic potential energy surface scans, and two-dimensional scans to parametrize in-ring torsions associated with the puckering states of dihydropterin and tetrahydropterin. The quality of the model was demonstrated through simulations of three protein complexes using optimized and initial parameters. These simulations underscored the significantly enhanced performance of the folate model developed in this study compared to the initial model without optimization in reproducing structural properties of folate–protein complexes. Overall, the presented MM model will be valuable for modeling folates in various redox states and serve as a starting point for parameterizing other folate derivatives.</p>\n </div>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2024-12-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27548","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
Folates comprise a crucial class of biologically active compounds related to folic acid, playing a vital role in numerous enzymatic reactions. One-carbon metabolism, facilitated by the folate cofactor, supports numerous physiological processes, including biosynthesis, amino acid homeostasis, epigenetic maintenance, and redox defense. Folates share a common pterin heterocyclic ring structure capable of undergoing redox reactions and existing in various protonation states. This study aimed to derive molecular mechanics (MM) parameters compatible with the CHARMM36 all-atom additive force field for pterins and biologically important folates, including pterin, biopterin, and folic acid. Three redox forms were considered: oxidized, dihydrofolate, and tetrahydrofolate states. Across all protonation states, a total of 18 folates were parameterized. Partial charges were derived using the CHARMM force field parametrization protocol, based on targeting reference quantum mechanics monohydrate interactions, electrostatic potential, and dipole moment. Bonded terms were parameterized using one-dimensional adiabatic potential energy surface scans, and two-dimensional scans to parametrize in-ring torsions associated with the puckering states of dihydropterin and tetrahydropterin. The quality of the model was demonstrated through simulations of three protein complexes using optimized and initial parameters. These simulations underscored the significantly enhanced performance of the folate model developed in this study compared to the initial model without optimization in reproducing structural properties of folate–protein complexes. Overall, the presented MM model will be valuable for modeling folates in various redox states and serve as a starting point for parameterizing other folate derivatives.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


