Further evidence of biallelic NAV3 variants associated with recessive neurodevelopmental disorder with dysmorphism, developmental delay, intellectual disability, and behavioral abnormalities.
{"title":"Further evidence of biallelic NAV3 variants associated with recessive neurodevelopmental disorder with dysmorphism, developmental delay, intellectual disability, and behavioral abnormalities.","authors":"Naseebullah Kakar, Selinda Mascarenhas, Asmat Ali, Azmatullah, Syed M Ijlal Haider, Vaishnavi Ashok Badiger, Mobina Shadman Ghofrani, Nathalie Kruse, Sohana Nadeem Hashmi, Jelena Pozojevic, Saranya Balachandran, Mathias Toft, Sajid Malik, Kristian Händler, Ambrin Fatima, Zafar Iqbal, Anju Shukla, Malte Spielmann, Periyasamy Radhakrishnan","doi":"10.1007/s00439-024-02718-6","DOIUrl":null,"url":null,"abstract":"<p><p>Neuron navigators (NAVs) are cytoskeleton-associated proteins well known for their role in axonal guidance, neuronal migration, and neurite growth necessary for neurodevelopment. Neuron navigator 3 (NAV3) is one of the three NAV proteins highly expressed in the embryonic and adult brain. However, the role of the NAV3 gene in human disease is not well-studied. Recently, five bi-allelic and three mono-allelic variants in NAV3 were reported in 12 individuals from eight unrelated families with neurodevelopmental disorder (NDD). Here, we report five patients from three unrelated consanguineous families segregating autosomal recessive NDD. Patients have symptoms of dysmorphism, intellectual disability, developmental delay, and behavioral abnormalities. Exome sequencing (ES) was performed on two affected individuals from one large family, and one affected individual from each of the other two families. ES revealed two homozygous nonsense c.6325C > T; p.(Gln2109Ter) and c.6577C > T; p.(Arg2193Ter) and a homozygous splice site (c.243 + 1G > T) variants in the NAV3 (NM_001024383.2). Analysis of single-cell sequencing datasets from embryonic and young adult human brains revealed that NAV3 is highly expressed in the excitatory neurons, inhibitory neurons, and microglia, consistent with its role in neurodevelopment. In conclusion, in this study, we further validate biallelic protein truncating variants in NAV3 as a cause of NDD, expanding the spectrum of pathogenic variants in this newly discovered NDD gene.</p>","PeriodicalId":13175,"journal":{"name":"Human Genetics","volume":" ","pages":"55-65"},"PeriodicalIF":3.6000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11754320/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00439-024-02718-6","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/12/21 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
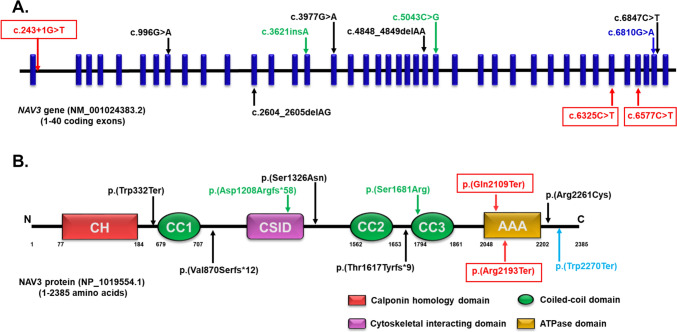
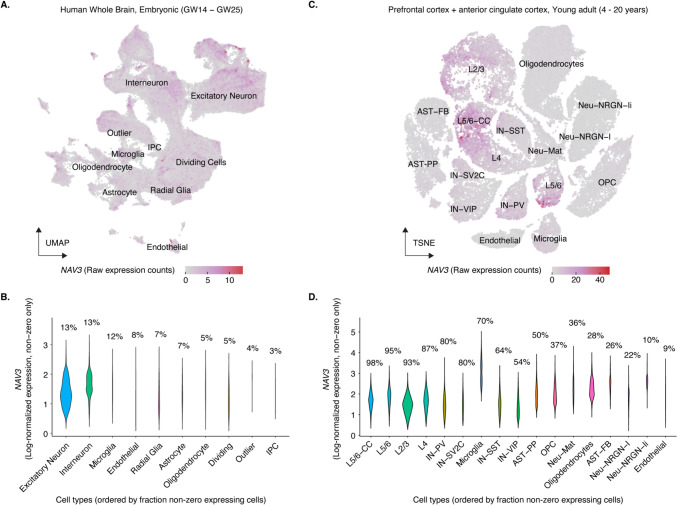
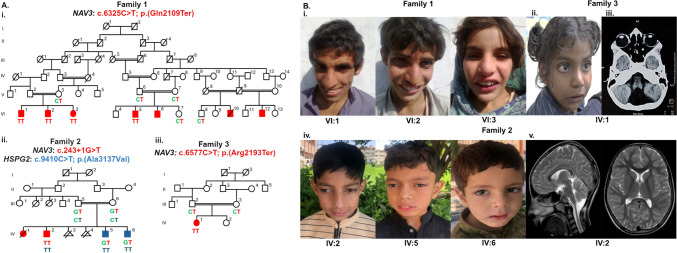
Neuron navigators (NAVs) are cytoskeleton-associated proteins well known for their role in axonal guidance, neuronal migration, and neurite growth necessary for neurodevelopment. Neuron navigator 3 (NAV3) is one of the three NAV proteins highly expressed in the embryonic and adult brain. However, the role of the NAV3 gene in human disease is not well-studied. Recently, five bi-allelic and three mono-allelic variants in NAV3 were reported in 12 individuals from eight unrelated families with neurodevelopmental disorder (NDD). Here, we report five patients from three unrelated consanguineous families segregating autosomal recessive NDD. Patients have symptoms of dysmorphism, intellectual disability, developmental delay, and behavioral abnormalities. Exome sequencing (ES) was performed on two affected individuals from one large family, and one affected individual from each of the other two families. ES revealed two homozygous nonsense c.6325C > T; p.(Gln2109Ter) and c.6577C > T; p.(Arg2193Ter) and a homozygous splice site (c.243 + 1G > T) variants in the NAV3 (NM_001024383.2). Analysis of single-cell sequencing datasets from embryonic and young adult human brains revealed that NAV3 is highly expressed in the excitatory neurons, inhibitory neurons, and microglia, consistent with its role in neurodevelopment. In conclusion, in this study, we further validate biallelic protein truncating variants in NAV3 as a cause of NDD, expanding the spectrum of pathogenic variants in this newly discovered NDD gene.
期刊介绍:
Human Genetics is a monthly journal publishing original and timely articles on all aspects of human genetics. The Journal particularly welcomes articles in the areas of Behavioral genetics, Bioinformatics, Cancer genetics and genomics, Cytogenetics, Developmental genetics, Disease association studies, Dysmorphology, ELSI (ethical, legal and social issues), Evolutionary genetics, Gene expression, Gene structure and organization, Genetics of complex diseases and epistatic interactions, Genetic epidemiology, Genome biology, Genome structure and organization, Genotype-phenotype relationships, Human Genomics, Immunogenetics and genomics, Linkage analysis and genetic mapping, Methods in Statistical Genetics, Molecular diagnostics, Mutation detection and analysis, Neurogenetics, Physical mapping and Population Genetics. Articles reporting animal models relevant to human biology or disease are also welcome. Preference will be given to those articles which address clinically relevant questions or which provide new insights into human biology.
Unless reporting entirely novel and unusual aspects of a topic, clinical case reports, cytogenetic case reports, papers on descriptive population genetics, articles dealing with the frequency of polymorphisms or additional mutations within genes in which numerous lesions have already been described, and papers that report meta-analyses of previously published datasets will normally not be accepted.
The Journal typically will not consider for publication manuscripts that report merely the isolation, map position, structure, and tissue expression profile of a gene of unknown function unless the gene is of particular interest or is a candidate gene involved in a human trait or disorder.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


