Understanding the Origin of the Redox Potential Shift of Transition Metal in LiFexMn1–xPO4 Cathodes by the Molecular Orbital Theory
IF 3.3
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
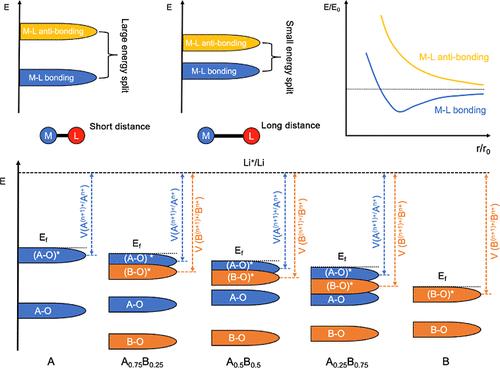
Olivine-structured LiFexMn1–xPO4 cathodes exhibiting higher redox potentials than their layer oxide counterparts have been utilized in commercial lithium-ion batteries, but the origin of the systematical shifts of the redox potential of transition metal couples with the variation of the Fe–Mn molar ratio is not clear, at least on the electronic scale. In the current work, we carried out experiments and theoretical calculations to study the molecular orbital characteristics of metal–ligand and determined the origin of transition metal redox potential shifts in LiFe1–xMnxPO4 cathodes on the electronic scale. The systematic shifts of redox potential of Fe3+/Fe2+ and Mn3+/Mn2+ couples in LiFe1–xMnxPO4 cathodes are not only because of the decreased energies of eg* antibonding orbitals with regard to the enlarged metal–ligand atomic distances but also due to almost the same slopes of the eg* antibonding orbital energies as a function of atomic distance. This chemistry picture of the metal–ligand atomic distance-dependent eg bonding/eg* antibonding splitting provides a new perspective to understand the redox potential variations of the electrode upon element substitution.
用分子轨道理论理解LiFexMn1-xPO4阴极过渡金属氧化还原电位移位的原因
具有较高氧化还原电位的橄榄石结构LiFexMn1-xPO4阴极已被用于商用锂离子电池中,但过渡金属偶对氧化还原电位随Fe-Mn摩尔比变化的系统变化的起源尚不清楚,至少在电子尺度上是如此。在目前的工作中,我们通过实验和理论计算研究了金属配体的分子轨道特征,并在电子尺度上确定了LiFe1-xMnxPO4阴极中过渡金属氧化还原电位位移的来源。LiFe1-xMnxPO4阴极中Fe3+/Fe2+和Mn3+/Mn2+对氧化还原电位的系统位移不仅是由于eg*反键轨道能量随金属-配体原子距离的增大而降低,而且由于eg*反键轨道能量随原子距离的变化斜率几乎相同。这张金属配体原子距离相关的eg键/eg*反键分裂的化学图为理解元素取代后电极的氧化还原电位变化提供了新的视角。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


