Accurate Singlet–Triplet Excited States Energy Gap Can Be Mastered by Time-Dependent Density Functional Theory Calculations Based on a Dielectric-Screened Range-Separated Hybrid Functional
IF 3.3
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
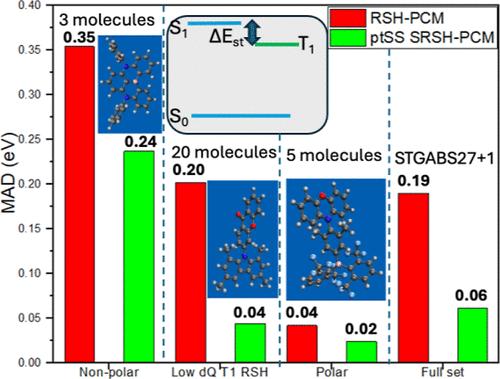
The energy gap between the lowest singlet and lowest triplet excited states of molecular emitters is a key property affecting their thermally activated delayed fluorescence (TADF) functionality. In cases in which these states are marked by internal charge transfer, the molecular environment of the emitter significantly stabilizes these states. To address this challenging relationship, a recently developed density functional theory (DFT) framework that alleviates the fundamental orbital gap in the gas phase can be employed, offering a highly effective and predictive description of these states. These modern functionals, range-separated hybrid functionals, are based on a generalized Kohn–Sham exchange functional expression where electronic interactions are separated into long and short ranges. For representing the significant condensed phase effects, especially in the case of charge transfer states, dielectric screening can be affected by functional expression. Dielectric-screened range-separated functionals that are invoked within a polarizable continuum model were shown to properly address the orbital gap challenge in the condensed phase and consequently benchmark well in calculating triplet and charge transfer excited states. We address the excited singlet–triplet state energy gap using such a screened range-separated functional and find that the calculated gaps tend to fall within 0.1 eV of the measured energies for a widely addressed benchmark set. Here, we emphasize the success of the dielectric-screened DFT framework in calculating these important energy gaps, reproducing well the benchmark values and therefore alleviating concerns about the applicability of existing functionals. We posit that dielectric-screened calculations will bear increasing impact on the design of organic materials aiming to enhance optoelectronic applications and in particular of realized TADF efficiencies.
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


